Альфа и бетта цепи гемоглобина

Что такое талассемия?
Талассемия (анемия Кули) — это группа наследственных расстройств, которые влияют на количество гемоглобина, производимых человеком. Гемоглобин относится к семейству соединений, состоящих из гема (железосодержащий комплекс) и различных глобинов (белковые цепи, которые окружают гемовый комплекс).
Молекулы гемоглобина (Hb) находятся во всех эритроцитах и являются причиной их красного цвета. Они связывают кислород в легких, переносят его через кровоток и выделяют в ткани организма. Различные типы гемоглобина классифицируются в соответствии с типом белковых цепей, которые они содержат.
Нормальные гемоглобины взрослых включают в себя:
- Гемоглобин А (составляет около 95% – 98% взрослого Hb). HbA содержит две альфа (α) белковые цепи и две бета (ß) цепи.
- HbA2 (составляет около 2% – 3,5% взрослого Hb), имеет две альфа (α) и две дельта (δ) цепи.
- HbF (до 2%). Это основной гемоглобин, вырабатываемый плодом во время беременности. Его производство обычно падает до низкого уровня в течение года после рождения. HbF имеет две альфа (α) и две гамма (γ) цепи.
Мутации в генах, кодирующих глобиновые цепи, могут вызывать нарушения в выработке гемоглобина. Есть 4 гена, которые кодируют цепи альфа-глобина, и 2 гена, которые кодируют цепи бета-глобина.
Наследственные нарушения производства гемоглобина делятся на две категории:
- Талассемия: снижение выработки нормальных гемоглобинов
- Гемоглобинопатия: производство аномальной молекулы гемоглобина
Талассемии представляют собой группу нарушений, при которых мутации в одном или нескольких из генов альфа- или бета-глобина вызывают уменьшение количества продуцируемого HbA. Это приводит к снижению уровня HbA, относительному увеличению количества минорных гемоглобинов HbA2 и HbF и, возможно, обнаружению необычных типов гемоглобина.
Талассемии обычно классифицируются по типу глобиновой цепи, синтез которой снижается.
Классификация талассемии
Альфа-талассемия
Альфа-талассемия возникает вследствие делеции или мутации в одной, или нескольких копиях гена 4-альфа-глобина. Чем больше затронутых генов, тем меньше вырабатывается альфа-глобина. Четыре различных типа альфа-талассемии включают в себя:
- Черта альфа-талассемии (1 пораженный ген). Безмолвный носитель будет иметь нормальные уровни гемоглобина и индексы эритроцитов, которые являются нормальными или демонстрируют слегка уменьшенный MCH (гипохромия). Носители могут передать пораженный ген своим потомкам. Часто эти люди идентифицируются только после рождения ребенка с болезнью HbH или признаком альфа-талассемии.
- Черта альфа-талассемии (2 пораженных гена). Пациенты с признаком альфа-талассемии имеют меньшие (микроцитарную), бледные (гипохромную) эритроциты и легкую хроническую анемию, но обычно они не испытывают никаких симптомов. Это анемия, которая не реагирует на добавки железа. Диагностика признака альфа-талассемии обычно происходит путем исключения других причин микроцитарной анемии. Подтверждающее тестирование с помощью анализа ДНК является важной частью процесса диагностики альфа-талассемии, поскольку на каждой хромосоме имеется две копии (HbA1 и HbA2) гена альфа-цепи. Если человек потерял обе копии из одной хромосомы, это имеет различные последствия для передачи более серьезной формы заболевания своим детям, чем если бы они потеряли одну копию из каждой двух разных хромосом. Смотрите раздел об анализе ДНК ниже.
- Болезнь гемоглобина Н (3 пораженных гена). При этом условии большое уменьшение количества производимых цепей альфа-глобина вызывает избыток бета-цепей, которые затем агрегируют в тетрамеры бета4 (группы из 4 бета-цепей), известные как гемоглобин H. Болезнь HbH может вызывать анемию от средней до тяжелой степени и спленомегалию (увеличенная селезенка). Клиническая картина, связанная с заболеванием HbH, чрезвычайно разнообразна. У некоторых людей она протекает бессимптомно, в то время как другие имеют тяжелую анемию. Болезнь гемоглобина Н чаще всего встречается у лиц юго-восточного или восточно-средиземноморского происхождения.
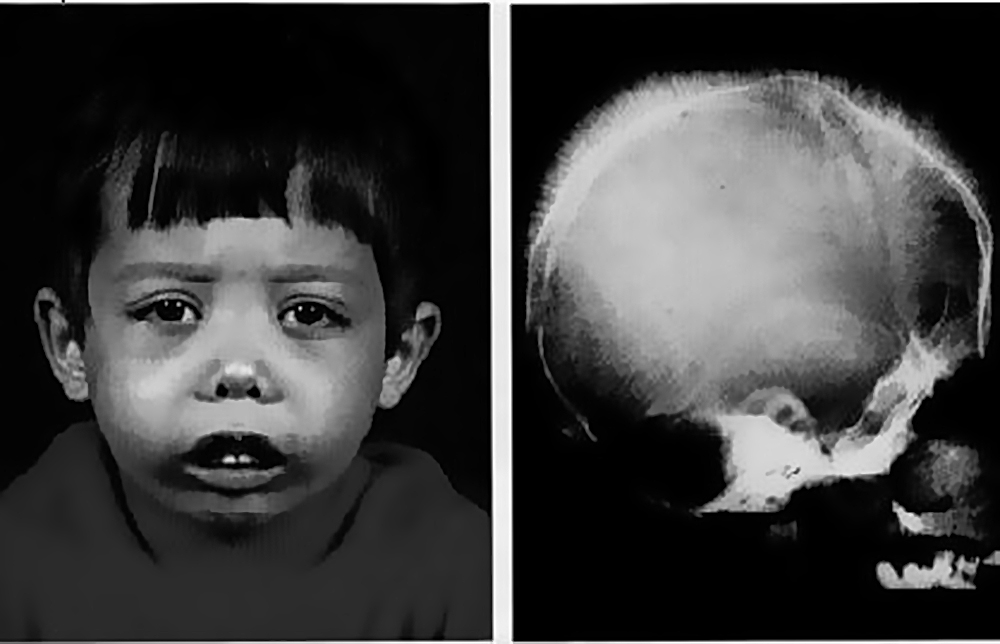
- Большая альфа-талассемия (также называемая фетальный гидропс, 4 пораженных гена). Это самая тяжелая форма альфа-талассемии. В этих условиях альфа-глобин не вырабатывается, следовательно, HbA или HbF также не вырабатываются. Плоды, пораженные альфа-талассемией, становятся анемичными на ранних сроках беременности. Они становятся отечными (сохраняют жидкость), и часто имеют увеличенные сердца и печень. Этот диагноз часто ставится в последние месяцы беременности, когда УЗИ плода указывает на отек плода. Приблизительно в 80% случаев у матери возникает токсемия и может развиться тяжелое послеродовое кровотечение. Дети с большой альфа-талассемией обычно недоношенные, мертворожденные или умирают вскоре после рождения.
Люди с альфа-талассемией могут быть неправильно диагностированы неосторожными врачами как дефицит железа, так как дефицит железа также приводит к появлению небольших бледных (микроцитарных гипохромных) эритроцитов.
Важно, чтобы лечение железом у больных талассемией назначалось только тогда, когда специфические анализы на железо (ферритин, сывороточное железо, TIBC, трансферрин) подтвердили дефицит железа. Это особенно важно при альфа-талассемии, где существует небольшая вероятность развития опасной перегрузки железом.
Альфа-талассемия чаще всего встречается у людей с этническим происхождением из Юго-Восточной Азии, Южного Китая, Ближнего Востока, Индии, Африки и Средиземноморья.
Бета-талассемия
Бета-талассемия происходит из-за мутаций в одном или обоих генах бета-глобина. Было выявлено от 100 до 200 мутаций, но распространены только около 20.
Тяжесть анемии, вызванной бета-талассемией, зависит от того, какие мутации присутствуют, и от того, уменьшают ли они выработку бета-глобина (так называемая бета + талассемия) или полностью ее устраняют (так называемая бета-талассемия). Различные типы бета-талассемии следующие:
- Бета-талассемия с характерной чертой. У человека с этим заболеванием один нормальный ген и один с мутацией. Обычно они не испытывают никаких проблем со здоровьем, кроме микроцитоза (маленьких эритроцитов) и возможной легкой анемии, которая не будет реагировать на добавки железа. Эта генная мутация может передаваться детям индивидуума.
- Талассемия интермедиа. В этом состоянии у больного человека есть два ненормальных гена, но он все еще производит некоторое количество бета-глобина. Тяжесть возникшей анемии и проблемы со здоровьем зависят от имеющихся мутаций. Разграничительной линией между промежуточной талассемией и большой талассемией является степень анемии, а также количество и частота переливаний крови, необходимых для ее лечения. Тем, у кого промежуточная талассемия, иногда требуется переливание, но не на регулярной основе.
- Талассемия крупная. Это самая тяжелая форма бета-талассемии. У пациента есть два ненормальных гена, которые вызывают либо серьезное снижение, либо полное отсутствие продукции бета-глобина, предотвращая выработку значительных количеств HbA. Это состояние обычно появляется у ребенка после 3-месячного возраста и вызывает опасную для жизни анемию. Эта анемия требует регулярных переливаний крови в течение всей жизни и значительной постоянной медицинской помощи. Со временем эти частые переливания приводят к чрезмерному количеству железа в организме. При отсутствии лечения это избыточное железо может откладываться в печени, сердце и других органах и может привести к преждевременной смерти от недостаточности органов.
Другие формы талассемии возникают, когда ген бета-талассемии наследуется в сочетании с геном варианта гемоглобина. Наиболее важными из них являются:
- HbE – бета-талассемия. HbE является одним из наиболее распространенных вариантов гемоглобина, встречающихся преимущественно у людей юго-восточного азиатского происхождения. Если человек наследует один ген HbE и один ген бета-талассемии, комбинация провоцирует талассемию HbE-бета, которая вызывает умеренно тяжелую анемию, подобную промежуточной бета-талассемии.
- HbS – бета-талассемия или серповидноклеточная – бета-талассемия. HbS является одним из наиболее известных вариантов гемоглобина. Наследование одного гена HbS и одного гена бета-талассемии приводит к талассемии HbS-бета. Тяжесть состояния зависит от количества бета-глобина, продуцируемого бета-геном. Если бета-глобин не вырабатывается, клиническая картина практически идентична серповидноклеточной анемии.
Какие обследования, анализы нужны?
Общий клинический анализ крови (ОАК). ОАК — это анализ клеток и жидкости в крови. Помимо прочего, ОАК сообщит врачу, сколько эритроцитов в крови, сколько гемоглобина содержится в них, и поможет измерить размер и форму присутствующих эритроцитов. Эти переменные называются индексами эритроцитов и включают MCH (средний корпускулярный гемоглобин) и MCV (средний корпускулярный объем), как измерения содержания гемоглобина и размера эритроцитов.
Низкий MCH или MCV часто являются первым признаком талассемии. Если уровень MCH или MCV низкий, а дефицит железа исключен, человек может быть носителем талассемии. При дефиците железа трудно исключить талассемию.
Микроскопическое исследование мазка крови. При этом тесте тонкий микроскопический слой крови исследуют на предметном стекле под микроскопом. Количество и тип лейкоцитов, эритроцитов и тромбоцитов можно подсчитать вручную и оценить, являются ли они нормальными и зрелыми. Различные нарушения влияют на нормальное производство клеток крови. При талассемии эритроциты часто бывают микроцитарными (маленькими) с низким MCV. Они также могут:
- Быть гипохромным (бледный — указывает низкий гемоглобин, чем обычно).
- Варьироваться по размеру (анизоцитоз) и форме (пойкилоцитоз).
- Быть зародышем (не нормально в зрелом эритроците).
- Быть искаженными, выглядеть под микроскопом как яблочки.
Анализ железа. Анализ может включать: железо, ферритин, UIBC, TIBC и процентное насыщение трансферрина. Эти тесты измеряют различные аспекты хранения и использования железа в организме. Они помогают определить, является ли дефицит железа причиной и/или обострением анемии пациента. Одному или нескольким пациентам с талассемией также может быть назначено наблюдение за степенью перегрузки железом.
Оценка гемоглобинопатии (Hb). Этот тест измеряет тип и относительное количество гемоглобинов, присутствующих в эритроцитах. Гемоглобин А, состоящий из альфа- и бета-глобина, является основным нормальным типом гемоглобина, обнаруживаемым у взрослых. Больший процент HbA2 и/или HbF обычно наблюдается при признаке бета-талассемии. HbH может наблюдаться при альфа-талассемии, но только тогда, когда по меньшей мере два из четырех альфа-генов удалены или мутированы.
Анализ ДНК, Во многих случаях анализ ДНК не требуется, поскольку диагноз может быть поставлен по результатам вышеуказанных тестов. Анализ ДНК чаще всего используется в семьях, страдающих альфа-талассемией. Этот тест используется для изучения делеций и мутаций в альфа и реже генах, продуцирующих бета глобин.
Семейные исследования могут быть проведены, чтобы оценить статус носителя и типы мутаций, присутствующих в других членах семьи. Исследование ДНК является важным инструментом для установления точного диагноза талассемии. В частности, при альфа-талассемии важно знать, имеет ли человек с чертой альфа-талассемии два мутированных гена в одной хромосоме или по одному в каждой хромосоме.
Лечение талассемии
Большинство людей с талассемией не нуждаются в лечении. Всем людям с диагнозом талассемия, которые планируют семью, настоятельно рекомендуется обратиться за генетическим советом, чтобы понять последствия для своего потомства. Требуется, чтобы лаборатория провела генетическое исследование партнера, чтобы генетический консультант предоставил точную информацию о риске передачи затронутых генов своим детям и серьезности талассаземии или гемоглобинопатии, которая может развиться.
Пациенты с гемоглобином Н или промежуточной бета-талассемией будут испытывать различные степени анемии в течение всей жизни. Они могут вести относительно нормальную жизнь, но требуют регулярного мониторинга и иногда могут нуждаться в переливании крови.
Добавки с фолиевой кислотой часто назначаются для борьбы с анемией, но следует избегать приема препаратов железа, если дефицит железа не подтвержден более конкретными исследованиями.
Часто изменения в показателях эритроцитов наблюдаются, когда женщина беременна, это может иногда приводить к тяжелой анемии. Понимание влияния диагноза талассемии на беременность и возможных последствий для здоровья потомства пары является одной из наиболее важных причин для установления точного диагноза талассемии.
Больным с бета-талассемией обычно требуется переливание крови каждые 3-4 недели в течение всей жизни. Эти переливания помогают поддерживать гемоглобин в достаточно высокой концентрации, чтобы обеспечить организм кислородом и предотвратить нарушения роста и повреждения органов.
Частые переливания, однако, поднимают железо до токсичных уровней, что приводит к отложению железа в печени, сердце и других органах.
Регулярная хелатная терапия железом используется для снижения уровня железа в организме Это включает введение препарата, который связывается с железом и помогает вымывать его из организма через мочу. Также часто больным может требоваться спленэктомия (хирургическая операция по удалению селезёнки).
Дети с большой альфа-талассемией обычно рождаются недоношенными, мертворожденные или умирают вскоре после рождения. Лечение сосредоточено на выявлении состояния и прерывании беременности или наблюдении за осложнениями у матери.
Экспериментальные методы лечения, такие как переливание крови плода, были успешными в очень немногих случаях при рождении ребенка.
Видеозаписи по теме
Источник

Снижение фракции гемоглобина (Нb) может сопровождать многие заболевания крови, чаще всего это связано с наследственным заболеванием – талассемией. Фракции гемоглобина, которые различаются по аминокислотному составу, сродству к кислороду, углекислому газу, имеют 3 вида: А (Нb взрослых),Р (Нb плода), F (Нb новорожденного). На молекулярном уровне структура Нb состоит из цепей глобина, строение и количество которых нарушается при талассемии.
Что такое талассемия

Талассемия – наследственная гемоглобинопатия с изменением количественного состава альфа и бета глобиновых цепей, которые входят в структуру гемоглобина А. При генетическом заболевании из-за нарушения строения гемоглобина разрушается внешняя оболочка эритроцита, что приводит к гибели клеток, выходу ее содержимого в кровь, гемолитической анемии.
Патология альфа чаще встречается в странах Западной Африки, Южной Азии, бета в Средиземноморье, Индонезии, Северной Африке, где часто встречается малярия. Согласно статистике, в мире каждый год рождается около 300 тыс. детей с разными видами талассемии. Признаки патологии могут отсутствовать в одном поколении, в другом привести к летальному исходу.
Важно!
Особенность талассемии в том, что она может выступать в виде защитного механизма против малярийного плазмодия.
Причины
Аномалия строения гемоглобина относится к наследственным аутосомно-рецессивным заболеваниям. В гене происходят мутации с изменением количества его цепей или их последовательности, которые влияют на синтез цепочек гемоглобина. На молекулярном уровне причиной является появление дефектной матричной рибонуклеиновой кислоты (РНК), нарушение транскрипции.
Читайте также
За счет этих нарушений синтез глобиновых цепей нарушается или полностью останавливается, формируется дефектный гемоглобин. Возможные причины молекулярных мутаций:
- вирусные болезни;
- влияние на организм химических мутагенов;
- наркотики;
- алкоголизм;
- противоопухолевые препараты;
- ионизирующая радиация.
Патология наследуется от родителей гомозиготно (получение мутированного гена от обоих родителей), гетерозиготно (дефектный ген достается ребенку от одного родителя).
Симптомы

Если мутированный ген унаследовался от одного родителя (гетерозиготно), симптоматика отсутствует либо болезнь проявляется слабо выраженными признаками в виде легкой анемии. Ее основные проявления – усталость, бледные кожные покровы. При диагностике выявляют незначительное увеличение селезенки, гипохромную анемию. Выраженные признаки заболевания наблюдаются, если дефектный ген передался от двух родителей (гомозиготно).
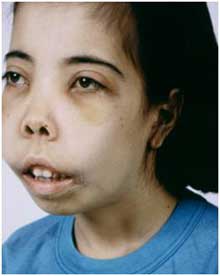
Чаще встречается бета-форма талассемии. Ее признаки проявляются в первый год жизни ребенка:
- изменение прикуса;
- переносица в виде седла;
- монголоидное лицо;
- изменение формы черепа (башенный, четырехугольный);
- увеличение печени, селезенки;
- землянисто-желтушный цвет кожи;
- патологические переломы костей при отсутствии травм;
- отставание в физическом развитии.
Возможно появление камней в желчном пузыре, воспаление суставов, язвенное поражение кожи ног. При талассемии разрушаются эритроциты, в крови повышается уровень железа, который может скапливаться в органах, вызывая цирроз печени, фиброзные изменения в поджелудочной железе, кардиосклероз, сахарный диабет.
Важно!
Альфа-талассемия, связанная с получением 2 дефектных генов от обоих родителей не совместима с жизнью, плод погибает внутриутробно.
Классификация
В зависимости от вида поврежденной цепи выделяют альфа-, бета, дельта – , гамма-талассемию. Последние 2 вида встречаются редко. Каждый вид включает в себя несколько подвидов.
Альфа-талассемия
Возникает из-за нарушения количества, структуры цепей альфа-глобина. Чем больше поврежденных генов, тем меньше синтезируется альфа-глобинов.
Альфа-талассемия с одним поврежденным геном
У человека с одним мутированным геном уровень эритроцитов, гемоглобина, как правило, в пределах нормы, возможна гипохромная анемия. Признаки заболевания отсутствуют, но человек носитель мутированного гена, который он может передать своим детям. Если ребенок унаследует только 1 поврежденный ген, он также станет носителем. Если он получит по одной поврежденной глобиновой цепи от каждого родителя, то проявятся характерные признаки талассемии.
Альфа-талассемия с двумя поврежденными генами

Симптоматика маловыражена или отсутствует. В крови определяют микроцитарные эритроциты, снижение количества здоровых эритроцитов. Проявляются признаки хронической анемии, выраженность которой не уменьшается на фоне лечения железосодержащими препаратами. Подтверждают генетическую микроцитарную анемию путем исключения других причин анемического состояния, ДНК-исследованиями с выявлением 2-х поврежденных цепей, которые могут быть повреждены в одном или двух генах.
Повреждение 3 генов – болезнь гемоглобина Н
Из-за повреждения трех альфа-цепей увеличивается количество бета-цепочек, которые замещают альфа. Заболевание вызывает анемию средней, тяжелой степени с увеличением размеров селезенки. Клиническая картина разнообразна, несмотря на серьезные нарушения в строении гемоглобина возможно бессимптомное носительство, но при передачи гена потомству возникают тяжелые нарушения в организме.
Повреждение 4 генов – большая альфа-талассемия
Самая тяжелая форма нарушения альфа-глобина. При таком повреждении цепи не синтезируются, соответственно нарушено синтезирование нормального, фетального гемоглобина. В 80% случаев плод погибает внутриутробно. Рожденные с большой талассемией дети имеют аномалии развития органов, как правило, умирают в течение 2-3 лет.
Вета-талассемия

Связана с мутацией одного или нескольких генов, кодирующих бета-глобиновые цепи. Известно до 200 мутаций. Виды:
- Бета-талассемия с одним поврежденным геном – не проявляется характерными для патологии симптомами, возможно уменьшение размеров эритроцитов, анемия легкой степени.
- Промежуточная форма с двумя поврежденными генами – глобиновые цепи синтезируются, но их количество снижено. Проявляется анемией средней степени.
- Тяжелая форма, связанная с угнетением синтеза бета-цепей. Начинает проявляется с 3-х месяцев жизни. Вызывает анемию тяжелой степени.
Другие виды возникают, если мутированный ген бета-цепи сочетается с патологической формой гемоглобина. Наиболее значимыми являются 2 формы: НbЕ, НbS (серповидно-клеточная бета-талассемия).
Важно!
Тяжелая форма бета-талассемии требует регулярных гемотрансфузий, медикаментозного лечения на протяжении всей жизни.
Диагностика
В постановке диагноза важен семейный анамнез, из которого можно узнать о случаях талассемии в роду. При физикальном исследовании обращают внимание на изменение строения черепа, костей носа, нижней челюсти, цвет кожи. При лабораторной диагностике при расшифровке общего, биохимического анализа крови для талассемии характерно:
- снижение уровня гемоглобина;
- изменение цветового показателя;
- высокий уровень непрямого билирубина;
- гипохромная анемия;
- эритроциты в форме мишени.
Чтобы определить фракции гемоглобина, содержание в крови его дефектных форм проводят электрофорез. При рентгенологическом исследовании черепа, трубчатых костей выявляют нарушения строения, формы костной ткани. При УЗИ возможно обнаружение камней в желчном пузыре, увеличение размеров печени, селезенки. Чтобы выявить мутированные гены проводят молекулярно-генетические исследования.
Читайте также
Лечение
Если клинические признаки талассемии не проявляются, лечение не требуется, чаще это касается бета-форм с наследованием гена от одного родителя. При рождении больного ребенка с двумя дефектными генами (по одному от каждого родителя) лечение требуется с первого месяца. Консервативная терапия включает в себя регулярные гемотрансфузии. Частоту процедур определяют по лабораторных показателям, выраженности симптомов, возрастным особенностям.
Для купирования периодов обострений талассемии назначают глюкокортикостероиды. Чтобы снизить уровень железа в крови показаны хелатирующие препараты. При талассемии снижается иммунитет, дети часто болеют инфекционными болезнями с осложнениями, поэтому назначают иммуномодуляторы, витаминные комплексы, вакцины против пневмококка.
Оперативное вмешательство показано при камнях в желчном пузыре, увеличение объема селезенки, которая сдавливает соседние органы. К хирургическому лечению относят пересадку костного мозга, если нарушается выработка форменных элементов. Для пересадки костного мозга подбирают только гистосовместимого донора.
Если верить статистике, примерно 1,5% мирового населения являются носителями β-талассемии без клинических симптомов. Профилактические мероприятия включают предупреждение браков между двумя носителями мутированных генов, проведение медико-генетических исследований при планировании ребенка, в период беременности. Для выявления нарушений строения гемоглобина необходимо дородовое инвазивное исследование (биопсия, забор амниотической жидкости).
Источник