Анализ крови фенилаланин норма

Медицинский эксперт статьи

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Референтные величины (норма) концентрации фенилкетонов в крови у детей – до 121 мкмоль/л (до 2 мг%).
Нарушение метаболизма фенилаланина относится к весьма распространённым врождённым расстройствам метаболизма. Вследствие дефекта гена фенилаланин гидроксилазы (ФАГ-ген) развивается недостаточность фермента, и как следствие наступает блок в нормальном превращении фенилаланина в аминокислоту тирозин. Количество фенилаланина в организме накапливается, и концентрация его в крови увеличивается в 10-100 раз. Далее он превращается в фенилпировиноградную кислоту, оказывающую токсическое воздействие на нервную систему. В связи с этим большое значение имеет ранняя диагностика фенилкетонурии, так как длительное существование фенилкетонемии приводит к нарушению умственного развития ребёнка. Накопление фенилаланина в организме происходит постепенно и клиническая картина развивается медленно.
Исследование крови проводят в течение ближайших 48 ч (2-5 дней), после того как новорождённый получил молоко (источник фенилаланина). На чашку с питательной средой, засеянной бактериями фенилаланин-зависимого штамма Bacillus subtilis, накладывают диск из фильтровальной бумаги, смоченной несколькими каплями капиллярной крови, и контрольные диски, содержащие разное количество фенилаланина. Зона роста бактерий вокруг диска, смоченного кровью, пропорциональна концентрации фенилаланина в крови новорождённого.
Помимо дефекта фенилаланин гидроксилазы к гиперфенилаланинемии может приводить транзиторная тирозинемия новорождённых, которая, вероятно, возникает в результате неадекватного метаболизма тирозина.
Типы гиперфенилаланинемии
Тип | Концентрация фенилаланина в крови, мг% | Дефектный фермент | Лечение |
Классическая фенилкетонурия | >20 | Фенилаланин гидроксилаза | Диета |
Атипичная фенилкетонурия | 12-20 | Фенилаланин гидроксилаза | Диета |
Персистирующая лёгкая гиперфенил-аланинемия | 2-12 | Фенилаланин гидроксилаза | Диета |
Транзиторная тирозинемия | 2-12 | Недостаточность β-гидроксифен-илпируват диоксигеназы (и др.) Вторичная в результате недостатка витамина С | Витамин С, смеси с низким содержанием белка |
Недостаточность дигидроптеридин редуктазы | 12-20 | Дигидроптеридин редуктаза | Допа, гидрокситриптофан |
Дефекты синтеза биоптерина | 12-20 | Дигидроптерид-инсинтетаза | Допа, гидрокситриптофан |
Транзиторная гиперфенил-аланинемия | 2-20 | Неизвестен | Нет |
Основа лечения пациентов с недостаточностью фенилаланингидроксилазы – ограничения фенилаланина в диете. При адекватно подобранной диете концентрация фенилаланина в крови не должна превышать верхний нормальный уровень или быть слегка ниже нормы.
У пациентов с транзиторной тирозинемией диета подбирается таким образом, чтобы концентрация тирозина в крови находилась в пределах между 0,5 и 1 мг%.
[1], [2], [3], [4], [5], [6], [7], [8]
Источник
Информация об исследовании
Фенилкетонурия – врожденное, передающееся по наследству нарушение обмена веществ. Причина – недостаточность фермента фенилаланингидроксилазы, необходимой для нормального метаболизма аминокислот, из которых состоят белки. В отсутствие этого фермента не происходит превращения аминокислоты фенилаланина в аминокислоту – тирозин. В результате резко возрастают уровни фенилаланина в крови и фенилкетона – производного фенилаланина – в моче.
Ген, связанный с фенилкетонурией PAH кодирует аминокислотную последовательность белковой молекулы фермента фенилаланин-4-гидроксилазы, который катализирует реакцию превращения L-фенилаланина в тирозин. Фенилаланин является незаменимой аминокислотой для построения белков и, кроме того, служит предшественником тироидных гормонов щитовидной железы, адреналина и меланина. Дефект фермента фенилаланин-4-гидроксилазы вследствие мутаций в гене PAH приводит к метаболическому блоку: поступающий с пищей фенилаланин не метаболизируется, а накапливается в организме.
В сыворотке крови его концентрация достигает 0,1 – 0,2 г/л (при норме 0,01—0,02 г/л). Активация альтернативных путей распада фенилаланина приводит к образованию и накопление в тканях токсических продуктов его обмена (фенилпировиноградной, фенилмолочной и других кетоновых кислот).
Симптомы фенилкетонурии проявляются в раннем детстве и включают рвоту, шелушащуюся кожную сыпь, раздражительность и затхлый (“мышиный”) запах тела, обусловленный аномальным составом мочи и пота. Симптомы со стороны центральной нервной системы могут быть разными, обычно это навязчивые движения, поддергивания, судороги. Самое тяжелое осложнение заболевания – задержка психического развития, которая в отсутствие лечения практически неизбежна.
Фенилкетонурия наследуется как рецессивный признак, что означает обязательное присутствие дефектного гена – причины данного заболевания – как у отца, так и у матери ребенка. Болезнь развивается лишь в том случае, если ребенок унаследовал оба дефектных гена. Вероятность рождения больного ребенка в семье, где оба родителя – носители дефектного гена, при каждой беременности составляет примерно 1:4.
Анализы крови на фенилкетонурию должны проводиться у всех новорожденных еще в родильных домах. Если анализ обнаруживает высокий уровень фенилаланина, необходимы дальнейшие исследования для подтверждения диагноза. Важность предварительных анализов у новорожденных связана с тем, что проявления заболевания, особенно задержку психического развития, можно предотвратить диетой с малым количеством фенилаланина.
Высокая частота заболевания, тяжелые нарушения в психической деятельности, поражения головного мозга в отсутствии адекватной терапии обуславливают необходимость раннего проведения ДНК-диагностики фенилкетонурии.
Комплексное генетическое исследование в Лаборатории Гемотест «Фенилкетонурия» позволяет выявить мутации основного гена PAH, связанного с моногенным заболеванием – фенилкетонурией. Исследование мутаций данного гена может иметь диагностическое значение у больных с клиническими проявлениями заболевания, а также прогностическое значение с целью выявления носительства неблагоприятных мутаций у здоровых лиц, вступающих в брак и/или планирующих деторождение.
Показания к назначению анализа:
- выявление предрасположенности к фенилкетонурии,
- клинический диагноз фенилкетонурия,
- супружеские пары, планирующие деторождение (в том числе имеющие здорового ребенка),
- супружеские пары, имеющие ребенка с фенилкетонурией,
- супружеские пары, являющиеся кровными родственниками,
- родственники больных фенилкетонурией.
Специальной подготовки к исследованию не требуется. Необходимо следовать общим правилам подготовки к исследованиям.
Общие правила подготовки к исследованиям:
1. Кровь рекомендуется сдавать утром, в период с 8 до 11 часов, натощак (между последним приемом пищи и взятием крови должно пройти не менее 8-ми часов, воду можно пить в обычном режиме), накануне исследования легкий ужин с ограничением приема жирной пищи.
2. Накануне исследования (в течение 24 часов) исключить алкоголь, интенсивные физические нагрузки, прием лекарственных препаратов (по согласованию с врачом).
3. За 1-2 часа до сдачи крови воздержаться от курения, не употреблять сок, чай, кофе, можно пить негазированную воду. Исключить физическое напряжение (бег, быстрый подъем по лестнице), эмоциональное возбуждение. За 15 минут до сдачи крови рекомендуется отдохнуть, успокоиться.
4. Не следует сдавать кровь для лабораторного исследования сразу после физиотерапевтических процедур, инструментального обследования, рентгенологического и ультразвукового исследований, массажа и других медицинских процедур.
Источник
Скрининговое обследование для исключения врождённых “ошибок” метаболизма по типу аминоацидопатий (наследственные нарушения обмена аминокислот). Показано взрослым с тяжелым нарушением обмена веществ. Определяемые аминокислоты: аланин (ALA), аргинин (ARG), цитруллин (CIT), пролин (PRO), глицин (GLY), метионин (MET), орнитин (ORN), фенилаланин (PHE), тирозин (TYR), валин (VAL), лейцин (LEU) + изолейцин (ILEU), аспарагиновая кислота (ASP), глутаминовая кислота (GLU).
Синонимы русские
Наследственные нарушения обмена аминокислот (аминоацидопатии).
Метод исследования
Высокоэффективная жидкостная хроматография-масс-спектрометрия (ВЭЖХ-МС).
Единицы измерения
Мкмоль/л (микромоль на литр).
Какой биоматериал можно использовать для исследования?
Венозную кровь.
Как правильно подготовиться к исследованию?
- Исключить из рациона алкоголь в течение 24 часов до исследования.
- Не принимать пищу в течение 8 часов до исследования, можно пить чистую негазированную воду.
- Полностью исключить (по согласованию с врачом) прием лекарственных препаратов в течение 24 часов перед исследованием.
- Исключить физическое и эмоциональное перенапряжение в течение 30 минут до исследования.
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Актуальность рассмотрения нарушений обмена аминокислот определяется тем, что эта патология отражается в первую очередь на функции нервной системы и является одной из основных причин слабоумия. Знание таких патологий необходимо в практике неонатологов и генетиков для профилактики и коррекции олигофрении.
По возможности синтезироваться аминокислоты в организме делятся заменимые и незаменимые. К незаменимым аминокислотам относятся аргинин, валин, изолейцин, лейцин, метионин, фенилаланин, к заменимым аминокислотам – аланин, глицин, тирозин. При дефекте ферментов на разных этапах трансформации аминокислоты и продукты их превращения могут накапливаться и оказывать отрицательное воздействие на организм.
Различают первичные (врождённые) и вторичные (приобретенные) нарушения метаболизма аминокислот. Врождённые заболевания обусловлены дефицитом ферментов и/или транспортных белков, которые связанны с метаболизмом аминокислот. Приобретенные нарушения аминокислот связаны с заболеваниями печени, ЖКТ, почек, недостаточным или неадекватным питанием, новообразованиями.
В норме наибольшая скорость обмена аминокислот характерна для нервной ткани. Поэтому разнообразные наследственные нарушения обмена являются одной из причин изменения функционирования в первую очередь ЦНС.
К числу наиболее серьезных и достаточно распространенных нарушений обмена относятся аномалии метаболизма фенилаланина и тирозина. Причина фенилкетонурии – врождённый дефицит печеночной фенилаланин–4–гидроксилазы. Это приводит к нарушению концентрации в крови, возникает дефицит тирозиновых и триптофановых производных (меланина, катехоламинов, серотонина). При этом в крови и моче значительно увеличиваются концентрации фенилацетилглутамина, фенилпирувата, фенилацетата. В крови повышается концентрация веществ, которые практически полностью отсутствуют в норме (фенилэтиламин, фенилпируват, фениллактат). Это нейротоксические соединения, они нарушают метаболизм липидов в мозге. В сочетании с дефицитом нейромедиаторов (серотонина) этот механизм считается ответственным за прогрессирующее снижение интеллекта у больных и развитие фенилпировиноградной олигофрении.
Лейциноз (“болезнь кленового сиропа”) – заболевание обусловлено дефицитом дегидрогеназы разветвленных кетокислот, которая катализирует реакцию окислительного декарбоксилирования. В результате нарушается окисление оксикислот с разветвленной цепью – ОКРЦ, которые образуются при катаболизме аминокислот с разветвленной цепью (лейцин, изолейцин, валин). У больных моча имеет специфический запах кленового сиропа. При данном заболевании особенно патогенно накопление лейцина. Это истинно кетогенная аминокислота. Кетоновые тела играют большую роль в энергообеспечении мозга, особенно при гипогликемии. Нарушение обмена лейцина приводит к развитию умственной отсталости, судорогам, мышечной ригидности, летаргии, рвоте. Отмечаются гипогликемия и кетоацидоз. Основным методом лечения является специальная диета.
Тирозинозы – болезни нарушения обмена тирозина – имеют несколько генокопий и носят аутосомно-рецессивный и аутосомно-доминантный типы наследования, сцепленные с полом. Заболеваемость – 1/20000 населения. Наиболее распространенной формой заболевания признается альбинизм. Наиболее частый механизм заболевания – дефект медьсодержащего фермента меланобластов тирозиназы, блокирующего превращение тирозина в диоксифенилаланин, из которого образуется эпинефрин и меланин. У альбиносов белые кожа и волосы, розово-красные глаза, фотодерматит. Больные страдают фотобоязнью и плохо видят днем вследствие депигментации сетчатки. Нарушение тирозинового обмена приводит к повреждению печени и развитию цирроза.
Поскольку тирозинозы имеют много генокопий и в патогенезе прослеживаются дефекты разных ферментов метаболизма тирозина, то клинически выделяют и другие формы. Среди них наиболее известны тирозиноз Медеса, гипертирозинемия I и II типов, хоукинсурия. При них тирозинемия с тирозинурией часто сочетаются с печеночной и почечной недостаточностью. Хоукинсурия имеет аутосомно-доминантный тип наследования и характеризуется выраженным слабоумием. Ферментативные дефекты метаболизма тирозина могут сопровождаться нарушением продукции тиреоидных гормонов на основе аминокислоты тирозина. Например, дефект йодтирозиндейодиназы – один из механизмов наследственного гипотиреоза с кретинизмом.
Исследование помогает определить уровень аминокислот в крови, их производных, оценить состояние аминокислотного обмена, диагностировать или подтвердить (при наличии характерных симптомов) нарушения обмена аминокислот. В него входит определение 13 показателей: аланин, аргинин, цитруллин, глицин, метионин, орнитин, фенилаланин, тирозин, лейцин/изолейцин, валин, аспарагиновая кислота, глутаминовая кислота.
Когда назначается исследование?
- Сходные случаи заболевания в семье;
- случаи внезапной смерти в раннем возрасте в семье;
- необычный запах тела и/или мочи (“сладкий”, “мышиный”, “вареной капусты”, “потных ног” и др.);
- неврологические нарушения – нарушения сознания (летаргия, кома), различные типы судорожных приступов, изменение мышечного тонуса (мышечная гипотония или спастический тетрапарез);
- нарушения ритма дыхания (брадипноэ, тахипноэ, апноэ);
- нарушения со стороны других органов и систем (поражение печени, гепатоспленомегалия, кардиомиопатия, ретинопатия);
- изменения лабораторных показателей крови и мочи: нейтропения, анемия, метаболический ацидоз/алкалоз, гипогликемия/гипергликемия, повышение активности печеночных ферментов и уровня креатинфосфокиназы, кетонурии, аммониемии;
- тяжелые нарушения обмена веществ;
- дифференциальная диагностика нарушений азотистого обмена.
Что означают результаты?
Референсные значения*
| Компонент | Референсные значения, мкмоль/л |
| Аланин (ALA) | 72.39 – 528.10 |
| Аргинин (ARG) | 14.30 – 83.27 |
| Валин (VAL) | 56.87 – 236.35 |
| Глицин (GLY) | 55.52 – 368.36 |
| Лейцин (LEU) | 48.97 – 255.92 |
| Метионин (MET) | 5.42 – 29.51 |
| Орнитин (ORN) | 18.51 – 79.68 |
| Пролин (PRO) | 72.13 – 177.07 |
| Тирозин (TYR) | 16.25 – 83.25 |
| Фенилаланин (PHE) | 16.22 – 72.34 |
| Цитруллин (CIT) | 8.16 – 32.91 |
| Аспарагиновая кислота (ASP) | 13.58 – 149.29 |
| Глутаминовая кислота (GLU) | 19.96 – 177.08 |
* – исследование предназначено только для лиц старше 18 лет.
Повышение общего количества аминокислот наблюдается:
- при эклампсии, нарушении толерантности к фруктозе, диабетическом кетоацидозе, почечной недостаточности, синдроме Рейе.
Снижение общего количества аминокислот наблюдается:
- при голодании, гиперфункции коры надпочечников, длительной лихорадке, хорее Гентингтона, синдроме мальабсорбции при тяжелых заболеваниях желудочно-кишечного тракта, гиповитаминозе, нефротическом синдроме, ревматоидном артрите.
Скрининг позволяет исключить следующие аминоацидопатии:
- болезнь “с запахом кленового сиропа мочи” (лейциноз);
- цитруллинемия тип 1, неонатальная цитруллинемия;
- аргининосукциновая ацидурия (АСА) / недостаточность аргининосукцинатлиазы;
- недостаточность орнитинтранскарбамилазы;
- недостаточность карбамилфосфатсинтазы;
- недостаточность N-ацетилглютамат синтазы;
- некетотическая гиперглицинемия;
- тирозинемия тип 1;
- тирозинемия тип 2;
- гомоцистинурия / недостаточность цистатионин-бета-синтетазы;
- фенилкетонурия;
- аргининемия / недостаточность аргиназы.
Также рекомендуется
[06-225] Анализ крови на аминокислоты (32 показателя)
[06-016] Гомоцистеин
[06-192] Анализ крови на органические кислоты
[06-193] Анализ мочи на органические кислоты
[02-006] Общий анализ мочи с микроскопией осадка
Кто назначает исследование?
Неонатолог, педиатр, невропатолог, инфекционист.
Источник

«Помню, когда ей было три месяца, она лежала в своей маленькой корзинке на прогулочной палубе корабля. Пока мы путешествовали, я приносила ее сюда, чтобы она дышала утренним воздухом. Люди, прогуливающиеся по палубе, останавливались взглянуть на нее, и меня одолевала гордость, когда они говорили о ее необычной красоте и о разуме в ее глубоких голубых глазах», — так писала о своей первой дочери Кэрол — американская писательница Перл Бак (“The Child Who Never Grew”, 1950). Автор длительно вынашивала идею написать это произведение не только для того, чтобы выразить свою боль, но и помочь другим родителям, находящимся в подобной ситуации. Но можно сказать, что эта новелла стала, вероятно, первым описанием ребенка с далеко не редкой болезнью: в 1960 году Кэрол, сильно отстающей в развитии и обучающейся в специальной школе, поставили диагноз «фенилкетонурия».
Хотя все началось несколько раньше…
В 1934 году физиолог Асбьерн Феллинг, изучавший метаболические расстройства, определил причину необычного запаха мочи у двух норвежских детей с умственной отсталостью: виной тому был избыточный уровень одного из метаболитов фенилаланина — фенилпировиноградной кислоты. Год спустя британцем Пенроузом был предложен термин «фенилкетонурия», а также определен аутосомно-рецессивный тип передачи заболевания. Помимо этого, Пенроуз предложил лечебную диету, но она не была принята. Аналогичная идея, озвученная Джервисом и Бикелем несколько позже, уже в 50-х, стала и остается до сих пор краеугольным камнем в лечении ФКУ. В 60-х микробиолог Роберт Гатри предложил диагностический тест для определения гиперфенилаланинемии: в качестве индикатора он использовал колонии Bacillus subtilis, которым для роста необходим фенилаланин. В наши дни многие страны по всему миру включили тест Гатри (либо более новые тестовые системы, основанные на тандемной масс-спектрометрии) в программы неонатального скрининга, что позволило сразу же приступить к лечению новорожденных и избежать серьезных нарушений интеллекта. Последние 20 лет прошлого века пролили свет на генетическую природу ФКУ, а в конце первого десятилетия 21-го века была сформирована база данных мутаций гена фермента фенилаланингидроксилазы, являющихся причиной развития заболевания. Примерно в это же время были установлены генетические причины нарушения метаболизма тетрагидробиоптерина.
Итак…
Фенилкетонурия (ФКУ) — врожденное нарушение метаболизма фенилаланина, приводящее к избыточному накоплению в биологических жидкостях фенилаланина (гиперфенилаланинемии, ГФА) и его дериватов.
Наиболее часто (~ 97–98 %) развитие ФКУ обусловлено мутацией гена фенилаланингидроксилазы (ФАГ), локализованного на длинном плече 12 хромосомы, участке 12q22–q24.1, которая наследуется аутосомно-рецессивно. Данный фермент лимитирует реакцию превращения фенилаланина в тирозин, и уровень ГФА, и, соответственно, тяжесть заболевания напрямую зависят от его активности, которая определяется особенностями мутации гена.
В остальных ~ 2–3 % случаев ФКУ вызвана недостаточностью тетрагидробиоптерина, которая развивается из-за мутацией гена одного или нескольких ферментов, регулирующих его обмен (BH4-дефицитная ФКУ). BH4 является коферментом ФАГ, а также некоторых других энзимов, опосредующих синтез дофамина и серотонина (см. рис.1).
В МКБ-10 выделяют «классическую ФКУ» и «другие гиперфенилаланинемии».
«Классический» вариант заболевания дифференцируется по степени тяжести согласно уровню фенилаланина в крови (см. табл.1)
Таблица 1 | Классификация классической ФКУ по степени тяжести
| Форма ФКУ* | Уровень фенилаланина в крови, мкмоль/л | Уровень фенилаланина в крови, мг/дл |
| Легкая ГФА** (не ФКУ) | 120–600 | 2–10 |
| Умеренная (мягкая, средняя) | 600–1200 | 10–20 |
| Классическая (тяжелая) | >1200 | >20 |
ФКУ* — фенилкетонурия; ГФА** — гиперфенилаланинемия
Благодаря результатам генетических исследований была создана классификация, отражающая этиопатогенез ГФА и ФКУ (см. табл.2)
Таблица 2 | Этиопатогенетическая классификация фенилкетонурии и гиперфенилаланинемии
| Название | Причинный фермент |
| ФАГ*-зависимая ФКУ** | Фенилаланин-4-гидроксилаза |
| ГФА***, BH4****-дефицит, тип А (ФКУ, 3 типа) | 6-пирувоил-тетрагидроптерин синтаза |
| ГФА, BH4-дефицит, тип B | Гуанозинтрифосфат-циклогидролаза |
| ГФА, BH4-дефицит, тип C (ФКУ, 2 типа) | Дигидроптеридинредуктаза |
| ГФА, BH4-дефицит, тип D | Птерин-4-альфа-карбиноламиндегидратаза |
| ГФА, BH4-дефицит | Сепиаптеринредуктаза |
ФАГ* — фенилаланингидроксилаза; ФКУ** — фенилкетонурия; ГФА*** — гиперфенилаланинемия; BH4**** — тетрагидробиоптерин
Другие ГФА встречаются как при физиологических, так и при патологических состояниях. У новорожденных может быть транзиторное повышение уровня фенилаланина в крови до патологических значений ввиду незрелости ферментных систем печени или избыточного белкового питания матери, но, как правило, состояние это не длительно, а клинические проявление незначительны либо вовсе отсутствуют. Патологическая ГФА может сопровождать поражения печени различной этиологии и в этом случае будет имеет вторичный характер.
Патогенез
Фенилаланин является незаменимой аминокислотой, поступающей в организм человека преимущественно в составе белковых продуктов животного происхождения. Большая часть этой аминокислоты расходуется на синтез собственных белков организма, а оставшаяся часть — на синтез тирозина, что является главным путем катаболизма фенилаланина. Эта реакция регулируется ферментом ФАГ при участии кофермента BH4 (см. рис 1). Отсутствие данного энзима либо его малое количество (при ФКУ от 0 до 50 % нормальной активности фермента) приводит к накоплению фенилаланина и развитию клинической картины ФКУ различной степени тяжести. Не утилизированный фенилаланин катаболизируется по минорному пути с образованием токсичных продуктов (фенилацетата, фенилпирувата,фениллактата), а сниженное образование тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов (меланинов). Помимо участия в синтезе тирозина, BH4 является коферментом в реакциях образования ДОФА и серотонина.
Также на количество медиаторов ЦНС влияет и само количество фенилаланина. Дело в том, что в норме фенилаланин, а также тирозин (как уже было обозначено выше — предшественник дофамина, норадреналина и адреналина) и триптофан (предшественник серотонина) преодолевают гематоэнцефалический барьер при помощи переносчика больших нейтральных аминокислот LAT1. Возросший при ФКУ уровень фенилаланина может ингибировать LAT1, препятствуя поступлению иных субстратов в нейроны.
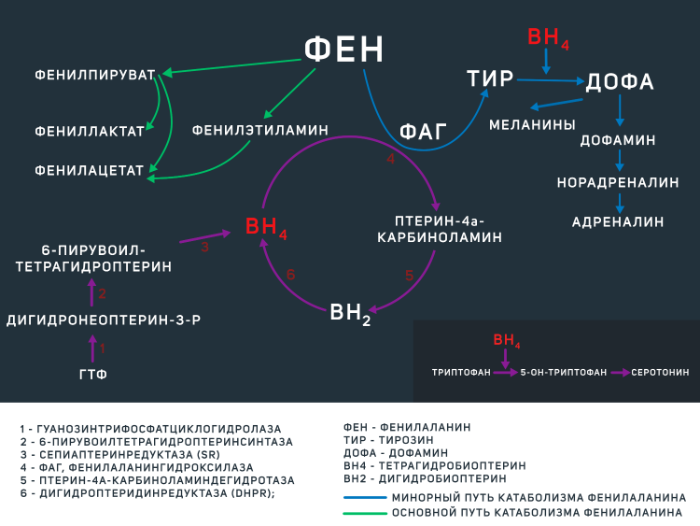
Рисунок 1 | метаболизм фенилаланина
Клиническая картина
Первые симптомы нелеченной ФАГ-зависимой ФКУ появляются, как правило, на первом году жизни ребенка, достигая максимума ко второму полугодию. Сперва обращает на себя внимание вялость ребенка либо, напротив, его беспокойство, возбужденность и срыгивания, нарушение мышечного тонуса, судороги, а также специфический затхлый запах мочи, названный «мышиным». Кроме того, нередко ФКУ проявляется эпилептическими приступами в виде абсансов, кивков, генерализованных судорог. Несколько позже, по мере роста ребенка, становится очевидным его задержка в моторном и нервно-психическом развитии. Болезнь, при отсутствии лечения, прогрессирует медленно, но неуклонно, приводя к глубокой олигофрении, несформированности речи, отсутствию игровой и предметной деятельности. Фенотипически для детей и взрослых, больных ФКУ, характерна гипопигментации кожи, волос и радужки.
При BH4-дефицитной ФКУ, помимо вышеобозначенных признаков, из-за большей недостаточности нейротрансмиттеров ЦНС выявляются атаксия, тремор, нарушения мышечного тонуса, гипокинезия, нарушения терморегуляции, затруднение глотания и поперхивания.
Диагностика
Первый этап лабораторной диагностики проводится на 3–7-й день жизни (но не ранее, чем через 2 дня от начала энтерального питания) новорожденного в рамках неонатального скрининга путем определения уровня фенилаланина на сухом пятне крови с помощью флюориметрии или тандемной масс-спектрометрии. При ГФА (фенилаланин > 120 мкмоль/л или > 2 мг/дл) проводится ретест. Если при повторном исследовании были получены подобные результаты, переходят ко второму этапу — определению отношения фенилаланин/тирозин. Этот косвенный метод позволяет провести дифференциальную диагностику между ФАГ-зависимой и BH4-зависимой ФКУ, что важно для назначения правильного лечения. Кроме лабораторных методов с целью уточнения типа заболевания используют молекулярно-генетические методы.
При отсутствие возможности провести неонатальный скрининг, в постановке диагноза опираются на клиническую картину, биохимические показатели, генеалогический анамнез, молекулярно-генетическую диагностику.
При выявлении легкой ГФА необходимо дальнейшее наблюдение и повторная диагностика.
Лечение
Основная цель терапии ФКУ — снижение уровня фенилаланина в крови для избежания нарушения моторного и нервно-психического развития ребенка — , достигается следующими методами:
- Гипофенилаланиновая диета — основной способ лечения уже более 60 лет. Для уменьшения поступления фенилаланина больным следует ограничивать прием высокобелковой пищи (мясо, рыба, яйца, молочные продукты, орехи, бобовые и др.) и вводить в рацион растительные продукты с высоким содержанием тирозина. Строгость диеты напрямую зависит от степени ГФА, меню должно составляться с опорой на факт «1 г белка = ~ 50 мг фенилаланина», возрастные физиологические нормы потребности в фенилаланине, тирозине и соотношение Б/Ж/У. У детей первого года жизни возможно употребление женского молока или молочных смесей при соответствующем расчете рациона и строгом контроле уровня фенилаланина в крови. Для восполнения недостающего белка используются аминокислотные смеси с низким содержанием фенилаланина и высоким содержанием тирозина, у детей старшего возраста компенсация происходит за счет растительных продуктов. Большой недостаток данного способа лечения — низкий комплаенс, особенно у детей подросткового возраста. Но при хорошей приверженности пациентов к диете снижение IQ можно свести к минимуму.
Некоторыми исследователями были получены данные об эффективности применения гликомакропептидов в диете. Гликомакропептиды (GLP, glycomacropeptides) — белки, получаемые из молочной сыворотки, которая богата валином, изолейцином, треонином и при этом содержит низкий уровень фенилаланина. Их использование позволило бы сделать гипофенилаланиновую диету более физиологичной, но для широкого применения необходимы дальнейшие исследования и подтверждение безопасности применения GLP в течение длительного срока. - Заместительная терапия BH4. Из-за участия BH4 в нескольких важных реакциях у больных BH4-зависимой формой ФКУ даже при хорошем соблюдении гипофенилаланиновой диеты остается симптоматика заболевания. В таком случае, как только на втором этапе лабораторной диагностики и/или на этапе медико-генетической диагностики подтверждается диагноз BH4-зависимой ФКУ, больным проводится тест на потенциальную чувствительность к сапроптерину дигидрохлориду — синтетическому аналогу BH4.
Иные методы лечения, имеющие потенциал:
- Большие нейтральные аминокислоты (The LNAAs, large neutral amino acids). Как было указано выше, фенилаланин способен конкурировать с другими аминокислотами (тирозин, триптофан) при взаимодействии с переносчиком LAT1. Некоторыми авторами было предположено, что в слизистой кишечника имеется подобный механизм, и при увеличении концентрации LNAAs всасывание фенилаланина будет уменьшаться.
- Генная терапия. Этот метод лечения мог бы стать идеальным решением, но в данный момент был тестирован лишь на мышах и требует дальнейшей серьезной разработки.
- Энзимотерапия фенилаланинамиаклиазой (PAL, phenylalanine ammonia-lyase). PAL — это фермент растений и дрожжевых грибков, осуществляющий катаболизм фенилаланина по альтернативному пути с образованием транс-циннамата и аммиака. За три последних десятилетия на мышах изучалось влияние PAL, внедренного в организм животного различными путями, начиная от оральных и инъекционных препаратов вплоть до помещения в кишечник генномодифицированных амеб, но, как и в случае с генной терапией, этот способ лечения требует дальнейшего изучения и разработки.
Источники:
- Blau N. et al. Phenylketonuria. // Lancet. Vol 376 October 23, 2010: pp 1417-1427.
- Blau N. Genetics of Phenylketonuria: Then and Now. // Human mutation, Vol 37, No. 6, 2016: pp 508-515.
- Hafid N.A., Christodoulou J. Phenylketonuria: a review of current and future treatments. // Translational Pediatrics 2015, 4(4): 304-317.
- Skirlou E., Lichter-Konecki U. Inborn Errors of Metabolism with Cognitive Impairment Metabolism Defects of Phenylalanine, Homocysteine and Methionine, Purine and Pyrimidine, and Creatine. // Pediatric Clinics of North America, Vol 65, 2018: pp 267-277.
- Руководство по педиатрии / [под ред. А.А. Баранова и др.] – Т: Врожденные и наследственные заболевания / [под ред. П.В.Новикова] – М.: “Династия”, 2007.
- Е.С. Северин и др.. Биологическая химия — М.: ООО «Медицинское информационное агентство», 2008.
- Клинические рекомендации “Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей”, 2017. https://www.pediatr-russia.ru/news/recomend
Источник