Анемия фанкони мкб 10

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Патогенез
- Симптомы
- Возможные осложнения
- Диагностика
- Лечение
- Список литературы
Другие названия и синонимы
Врожденная панмиелопатия Фанкони, Наследственная панмиелопатия.
Названия
Название: Анемия Фанкони.
Анемия Фанкони
Синонимы диагноза
Врожденная панмиелопатия Фанкони, Наследственная панмиелопатия.
Описание
Это генетическое заболевание, передающееся аутосомно-рецессивным путем, которое характеризуется нарушением кроветворения, образованием злокачественных опухолей, пороками развития и ломкостью хромосом. Это проявляется частым кровотечением, синяками, вялостью, бледностью и склонностью к инфекции. Диагностика проводится лабораторными методами, рекомендуются цитогенетические, генетические молекулярные и клинические анализы крови, миелограмма. Основными методами лечения являются трансплантация костного мозга, медицинское обеспечение кроветворения, переливание крови.
Анемия Фанкони
Дополнительные факты
Синонимами анемии Фанкони являются врожденная панмиелопатия Фанкони, наследственная панмиелопатия. Болезнь получила свое название от швейцарского педиатра Гвидо Фанкони, который в 1927 году описал врожденную апластическую патологию на основе симптомов трех братьев. Анемия Фанкони является редким генетическим заболеванием, наследуемым по аутосомно-рецессивному принципу. Эпидемиологические показатели невысоки: на 1 больного ребенка приходится 350 тысяч младенцев. Распространенность одинакова между представителями мужского и женского пола; оно выше в общинах с разрешенными близкими родственниками, например, среди некоторых южноафриканских народов.
Причины
Болезнь наследственная; это развивается, когда дефектный ген передается от родителей ребенку. Было идентифицировано 15 генов, мутации которых проявляются анемией Фанкони. Из них 14 расположены в аутосомах и являются рецессивными, 1 тип гена расположен в Х-хромосоме (связан с полом). Все эти гены отвечают за выработку определенного фермента, участвующего в репарации ДНК.
Аутосомно-рецессивное наследование подразумевает, что отец и мать должны нести патологическую генетическую информацию. Кроме того, они вообще здоровы. Вероятность рождения больного ребенка в такой паре составляет 25%. Генетическая панмиелопатия диагностируется у детей и взрослых, которые получают один и тот же измененный ген от каждого родителя. В очень редких случаях анемия вызвана передачей дефектной Х-хромосомы. Женщины могут нести мутации, болезнь проявляется только у мальчиков. Риск развития патологии у сына, если у матери мутированный ген, составляет 50%.
Патогенез
Обычно в клетках организма существуют специальные ферментативные системы, которые корректируют разрывы молекул ДНК, поврежденных при биосинтезе или воздействии химических, физических реагентов. При анемии Фанкони в кластере белков, ответственных за репарацию ДНК, обнаружен генетический дефект, что приводит к повышенной ломкости хромосом. В результате у пациентов развивается дисфункция костного мозга – рак и апластическая анемия. Онкологические заболевания чаще всего представлены острым миелоидным лейкозом – злокачественной опухолью костного мозга, которая вызывает накопление измененных лейкоцитов, которые ингибируют рост эритроцитов, тромбоцитов и нормальных лейкоцитов. При апластической анемии рост и созревание всех трех типов клеток крови быстро ингибируются дисплазией костного мозга.
Симптомы
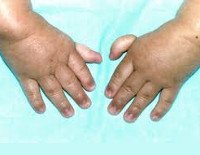
Более половины пациентов имеют врожденные аномалии внутренних органов и скелета. Деформации костей проявляются специфическим внешним видом: пациенты имеют низкорослый вид, уменьшенный размер головы, отсутствие или заметное укорочение большого пальца на руках, недоразвитие радиуса, врожденный вывих бедра и / или наличие шейного ребра, косолапость недоразвитый подбородок («птичье лицо»). Характерна гиперпигментация кожи в виде светлых и коричневатых пятен.
Неврологические расстройства представлены косоглазием, недоразвитием одного или двух глаз, опущением верхнего века, дрожанием глаз, глухотой, умственной отсталостью. Пациенты часто имеют незрелые половые органы, им не хватает одного или обоих яичек. Общие нарушения в структуре органов включают дефекты мочевыделительной системы: удвоение мочеточников или таза, подковообразные почки, почечные кисты, смещение наружного отверстия мочеиспускательного канала (гипоспадия). Врожденные пороки сердца включают атрезию трикуспидального клапана, дефект межпредсердной перегородки, митральный стеноз, дефект межжелудочковой перегородки. Пациенты страдают от почечной и сердечной недостаточности.
Основные симптомы связаны с постепенным увеличением нарушений костного мозга. Чаще всего они дебютируют в детстве (от 5 до 10 лет). Из-за уменьшения количества тромбоцитов усиливается кровотечение: кровь не сгущается в течение длительного времени, носовые кровотечения возникают легко, выделения во время менструации обильные, много «беспричинных» синяков на теле. Снижение количества эритроцитов проявляется анемией с характерной слабостью, быстрой утомляемостью, головокружением, обмороком, бледностью кожи, сердцебиением и одышкой. Дефицит лейкоцитов способствует ухудшению устойчивости к инфекциям. Впоследствии образуются лейкемия, миелодиспластический синдром и рак.
Вялость. Одышка.
Возможные осложнения
Частые инфекционные заболевания считаются наиболее распространенным осложнением. У пациентов развиваются ОРВИ, тонзиллит, ринит, бронхит, грипп, брюшной тиф и герпес. Рецидивирующий характер заболевания и его тяжелое течение приводят к разрушению органов, сопровождающемуся риском сепсиса. Еще одним осложнением наследственной анемии являются злокачественные новообразования – лейкемия, эпителиальные опухоли шеи и головы, половых органов. Рак у этих пациентов трудно поддается лечению из-за повышенной хрупкости и уменьшения восстановления ДНК. Это явление ограничивает применение лучевой терапии цитотоксическими препаратами. Нарушение свертываемости крови становится причиной большой потери крови.
Диагностика
Обследование пациентов проводится онкологами, гематологами, педиатрами и генетиками. Диагноз начинается с анализа анамнестических данных и жалоб. Врач выясняет, страдают ли эти родственники от этого наследственного заболевания, указывает время, в которое появляются первые признаки заболевания, ранние визиты к врачу. Во время обследования оценивается общее состояние пациента и выявляется наличие нарушений развития, гиперпигментированных пятен, кровотечений и кровоподтеков. В большинстве случаев нетрудно определить типичные деформации костей и недоразвитие глаз. Чтобы подтвердить диагноз и отличить анемию Фанкони от приобретенной анапластической анемии, проводится ряд лабораторных исследований:
• Клинический анализ крови. Изменения клеточного состава крови характерны. Тромбоцитопения и лейкопения диагностируются на ранних стадиях кроветворения, а панцитопения (резкое уменьшение объема эритроцитов, лейкоцитов и тромбоцитов) на поздних стадиях. Небольшой гемолиз возможен без гипербилирубинемии, но с ретикулоцитозом. Значение СОЭ возросло до 60-80 мм /.
• Цитогенетическое исследование клеток. Тест проводится с диэпоксибутаном, митомицином С, с указанием частоты и спектра хромосомных аберраций. В пользу генетической анемии параметры теста DEB считаются более 45%, пограничный уровень составляет 11-45% (процент клеток с хромосомными нарушениями).
• Молекулярно-генетический анализ клеток. Изучены гены, в которых мутации могут привести к развитию заболевания. В 60-70% случаев мутации обнаруживаются в паре генов FANCA, в 14% в аллеле FANCC и в 10% в генах FANCG. Уровень мутаций в других парах составляет 0,2-3%. Согласно исследованию, установлено увеличение количества фагоцитарных жиров в плазматических клетках и макрофагах. Содержание недифференцированных клеток находится в пределах нормы. Концентрация зародышевых клеток миелоцитов была снижена, а количество лимфоцитов увеличено.
Лечение
Основная терапия направлена на восстановление процесса кроветворения. Методы лечения подбираются индивидуально, в зависимости от тяжести заболевания, возраста пациента, наличия и тяжести врожденных дефектов. Кроме того, лечатся инфекции и онкопатологии и принимаются меры по реабилитации. Для устранения анемии используются следующие методы:
• BMT. Трансплантация костного мозга наиболее эффективна в отдаленном периоде, но имеет противопоказания, часто сопровождается развитием осложнений. Оптимальный возраст для операции – до десяти лет. Здоровые сестры и братья, которые соответствуют критериям отбора, могут быть донорами. Предварительная интенсивная терапия (кондиционирование) связана с риском токсического воздействия на органы. После трансплантации вероятность острого или хронического иммунного конфликта между донорскими и реципиентными клетками остается высокой.
• Медикаментозная стимуляция кроветворения. Если трансплантация невозможна, пациентам показано консервативное лечение, которое временно улучшает их состояние. Продукцию клеток крови стимулируют андрогены (мужские половые гормоны) и гематопоэтические факторы роста – эритропоэтин, фактор стволовых клеток, интерлейкины-1-12. Параллельно используются иммунодепрессанты. Медикаментозная терапия на протяжении многих лет поддерживала высокое качество жизни пациентов, но ее эффективность постепенно снижалась.
• Переливание компонентов крови. При тяжелых побочных эффектах или противопоказаниях к этиотропной терапии (трансплантация, стимуляция кроветворения) назначаются процедуры переливания крови. Промывают эритроциты, донорские эритроциты освобождаются от поверхностных белков, переливаются. При кровотечении и снижении уровня тромбоцитов пациентам вводят массу тромбоцитов.
Список литературы
1. Генетическая диагностика анемии Фанкони. Обзор литературы/ Панферова А. В. Тимофеева Н. М. Ольшанская Ю. В. // Онкогематология. – 2016.
2. Анемии (клиника, диагностика, лечение). Методическое пособие/ Филатов Л. Б. – 2006.
3. Свободнорадикальный статус клеток крови и костного мозга у детей с острыми лейкозами и анемией Фанкони: Автореферат диссертации/ Чивилева И. Ю. – 1996. V.
4. Анемия Фанкони: современная диагностика и терапия: Автореферат дисертации/ Кравченко Е. Г. – 2003.
- Описание
- Причины
- Симптомы (признаки)
- Диагностика
- Лечение
Краткое описание
Апластические анемии — группа патологических состояний, характеризующихся панцитопенией в периферической крови вследствие угнетения кроветворной функции костного мозга.
Код по международной классификации болезней МКБ-10:
- D60 Приобретенная чистая красноклеточная аплазия (эритробластопения)
- D61 Другие апластические анемии
Классификация • Врождённые (анемия Фанкони, Дайемонда–Блекфэна) • Приобретённые (результат воздействия химических, лекарственных, инфекционных агентов и ионизирующего излучения) • Идиопатические (причина заболевания неясна).
Причины
Этиология • Приобретённые анемии •• Инфекции (вирусные гепатиты, инфекционный мононуклеоз, ЦМВ, парвовирусная, грипп) •• Лимфопролиферативные нарушения (тимома, лимфома, хронический лимфобластный лейкоз) •• Ионизирующее излучение •• Токсины и химические вещества (бензол, инсектициды, соединения свинца и пр.) •• ЛС, например цитостатики, препараты золота, НПВС, противосудорожные препараты, антибиотики • Врождённые анемии (анемия Фанкони). Существует 4 группы комплементации, т.е. не менее 4 генов, мутация любого из которых ведёт к развитию апластической панцитопении 4 типов (все r) •• Группа А (тип 1 анемии Фанкони, *227650, 20q13.2–q13.3, дефекты генов FA1, FA, F, r) •• Группа В (тип 2, *227660, дефект гена FA2, r) •• Группа С (тип 3, *227645, 9q22.3, дефект гена FACC, r) •• Группа D (тип 4, *227646, дефект гена FA4, r).
Патогенез апластических анемий • Внутренний дефект гемопоэтических стволовых клеток • Иммунная реакция на гемопоэтическую ткань • Нарушение поддерживающей функции стромального окружения в костном мозге
Симптомы (признаки)
Клиническая картина
• Характерные черты апластических анемий •• Общие признаки (одышка, бледность кожных покровов и слизистых оболочек, тахикардия, слабость, систолический шум на верхушке сердца, снижение массы тела) •• Частое присоединение инфекционно – воспалительных и гнойно – некротических процессов различной локализации вследствие лейкопении (нейтропении) •• Геморрагический синдром: крупные и мелкие кровоизлияния (в т.ч. в сетчатку глаза), кровотечения различной локализации (меноррагия, мелена, носовые кровотечения), обусловленные тромбоцитопенией. Особенности у детей • Преобладающий возраст — 14–16 лет • Анемии обычно предшествует перенесённое инфекционное заболевание (ветряная оспа, краснуха, вирусные гепатиты) • Заболевание развивается очень быстро • В 70% случаев при лечении циклоспорином удаётся достичь полной клинико – гематологической ремиссии.
• Анемия Фанкони. Существует 2 типа •• Классический (тип I) ••• Низкий рост ••• Микроцефалия ••• Деформации скелета (отсутствие или гипоплазия первой пястной или лучевой кости) ••• Аномалии почек и сердца ••• Гипоспадия ••• Гиперпигментация кожи ••• Глухота •• Тип 2 характеризуется наличием малых аномалий развития.
• Врождённая апластическая анемия (синдром Дайемонда–Блекфэна) — аллельный вариант анемии Фанкони (*205900, r). От анемии Фанкони её отличают раннее появление анемии (как правило, в первые месяцы жизни), отсутствие нейтропении и тромбоцитопении.
По тяжести различают нетяжёлую (гранулоциты меньше 1,5´109/л, тромбоциты 100´109/л, мегакариоциты меньше 80´109/л) и тяжёлую (в периферической крови гранулоциты менее 0,5´109/л, тромбоциты менее 20´109/л; в костной ткани преобладание фиброзной ткани более чем на 60%) формы.
ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ
• Приобретённая анемия •• Панцитопения •• Нормохромные эритроциты •• Удлинённое время кровотечения •• Выраженное снижение содержания ретикулоцитов (арегенераторная анемия) •• Повышение концентрации железа в сыворотке крови (вследствие гемотрансфузий) •• Нормальные показатели общей железосвязывающей способности сыворотки (ОЖСС) •• Концентрация эритропоэтина сыворотки обычно увеличена пропорционально степени анемии •• Нарушение показателей функций печени.
• Анемия Фанкони •• Макроцитоз (в отличие от приобретённой апластической анемии) •• Повышение содержания HbF •• Отсутствие выраженной панцитопении до 3–8 – летнего возраста •• Характерны ломкость хромосом, дефекты репарации, повышенная чувствительность хромосом к диэпоксибутану, митомицину и УФО.
• Анемия Дайемонда–Блекфэна: макроцитоз, ретикулоцитопения, повышенное содержание HbF.
Пункция костного мозга • Увеличенные запасы железа • Уменьшено количество клеток •• Уменьшение количества мегакариоцитов •• Уменьшение количества миелоцитов •• Уменьшение количества эритроидных предшественников •• Замещение нормальной кроветворной ткани на жировую и фиброзную • Анемия Фанкони — при пункции костного мозга часто не обнаруживают изменений.
Диагностика
Инструментальные исследования • КТ области тимуса при подозрении на тимому • Рентгенологическое исследование костей предплечья (возможно обнаружение аномалий развития лучевой кости) и больших пальцев кистей (конституциональная анемия) • УЗИ почек.
Дифференциальная диагностика • Транзиторная эритробластопения у детей • Миелодиспластический синдром • Пароксизмальная ночная гемоглобинурия • Острый лейкоз • Волосатоклеточный лейкоз • СКВ • Диссеминированная инфекция • Гиперспленизм • Анемии при гипотиреозе, гипопитуитаризме и заболеваниях печени.
Лечение
ЛЕЧЕНИЕ
Тактика ведения • Стационарное лечение в гематологическом отделении. При нейтропении показана изоляция для предупреждения возможного инфицирования • Исключение причинных факторов • Исследование Аг системы HLA больных и членов их семей.
Трансплантация костного мозга пациентам с выраженной апластической анемией, особенно в возрасте до 30 лет — метод выбора (при наличии HLA – идентичного донора) • Трансплантация от неродственных доноров в случае неэффективности проводимого лечения.
Консервативное лечение
• Проводят при отсутствии донора для трансплантации костного мозга •• Глобулин антитимоцитарный. Для выявления повышенной чувствительности сначала необходимо проведение кожной пробы ••• Препарат выбора для лечения пациентов пожилого возраста или при отсутствии донора для трансплантации костного мозга ••• Курсовая доза 160 мг/кг, разделённая на 4 дозы, в течение первых 4 дней лечения ••• Необходимую дозу препарата растворяют в 500 мл 0,9% р – ра натрия хлорида и вводят в/в капельно в течение 4–6 ч ••• Развитие анафилактической реакции — абсолютное противопоказание к дальнейшему введению препарата •• Циклоспорин ••• Начальная доза — 5 мг/кг/сут внутрь или 3 мг/кг в/в. Далее дозы подбирают, исходя из концентрации циклоспорина в крови, определяемой ежедневно ••• При отсутствии эффекта в течение 120 дней препарат отменяют •• Метилпреднизолон — по 2 мг/кг/сут в/в с 1 по 14 день лечения, по 1 мг/кг — с 15 по 21 день •• Гранулоцитарный колониестимулирующий фактор (например, молграмостим) — при неэффективности тимоглобулина или циклоспорина ••• Начальная доза — 5 мкг/кг/сут п/к до увеличения количества гранулоцитов более 1 000/мкл ••• При отсутствии эффекта в течение 14 дней дозу удваивают •• Андрогены эффективны при некоторых вариантах анемии Фанкони, приобретённой апластической анемии, хотя случаи успешного лечения крайне редки. При отсутствии эффекта в течение 4–6 мес препарат отменяют постепенно.
• Поддерживающая терапия •• Оксигенотерапия •• Санация полости рта и других слизистых оболочек •• Переливание отмытых эритроцитов через лейкоцитарные фильтры; тромбоцитарной массы •• Антибактериальная терапия •• Гемостатики
• Осложнения терапии • Геморрагический синдром • Инфекционные осложнения • Гемосидероз при переливании крови • Сердечная недостаточность • Почечная недостаточность • Токсический гепатит • Анафилактический шок и сывороточная болезнь (при использовании антитимоцитарного глобулина) • Развитие острого лейкоза.
Оперативное лечение • Тимэктомия при тимоме.
Исходы лечения • Полная клинико – гематологическая ремиссия: концентрация Hb выше 110 г/л, тромбоцитов более 100´109/л, гранулоцитов более 1,5´109/л • Частичная клинико – гематологическая ремиссия: концентрация Hb 90–110 г/л, тромбоцитов 30–100´109/л, гранулоцитов 0,5–1,5´109/л • Минимальный гематологический ответ: уровень Hb 80–90 г/л, тромбоцитов более 10–30´109/л (без проведения трансфузий).
Течение и прогноз • Приобретённая анемия. Течение и прогноз прямо пропорциональны возрасту больного. При отсутствии своевременной и адекватной терапии 80% пациентов погибают в течение 3 мес от манифестации заболевания. С внедрением в клиническую практику циклоспорина шансы на продление жизни больного увеличились. При предполагаемой трансплантации костного мозга во избежание сенсибилизации в предоперационный период желательно избегать переливаний препаратов крови (в особенности от родственников) • Анемия Фанкони. Выживаемость не превышает 4 лет при изолированной заместительной терапии. Прогноз значительно благоприятнее при эффективности андрогенов или трансплантации костного мозга.
Синонимы: Для анемии Фанкони — синдром Фанкони.
МКБ-10 • D60 Приобретённая чистая красноклеточная аплазия [эритробластопения] • D61 Другие апластические анемии
Примечания • Аплазия костного мозга — врождённое или приобретённое тяжёлое состояние, проявляющееся панцитопенией. Врождённая форма сочетается с фенотипическими и цитогенетическими аномалиями, что облегчает диагностику • Панцитопения — снижение количества эритроцитов, лейкоцитов и тромбоцитов. Характерны бледность и сонливость, инфекции, обусловленные нейтропенией; геморрагический диатез вследствие тромбоцитопении. Для дифференциальной диагностики необходимо исследование костного мозга (аплазия [чаще гипоплазия] костного мозга, вытеснение ростков костного мозга бластными клетками, аутоиммунное разрушение клеток крови — синдром Эванса) • Конституциональными называют врождённые и иногда идиопатические типы анемий.