Анемия фанкони у детей клинические рекомендации

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Патогенез
- Симптомы
- Возможные осложнения
- Диагностика
- Лечение
- Список литературы
Другие названия и синонимы
Врожденная панмиелопатия Фанкони, Наследственная панмиелопатия.
Названия
Название: Анемия Фанкони.
Анемия Фанкони
Синонимы диагноза
Врожденная панмиелопатия Фанкони, Наследственная панмиелопатия.
Описание
Это генетическое заболевание, передающееся аутосомно-рецессивным путем, которое характеризуется нарушением кроветворения, образованием злокачественных опухолей, пороками развития и ломкостью хромосом. Это проявляется частым кровотечением, синяками, вялостью, бледностью и склонностью к инфекции. Диагностика проводится лабораторными методами, рекомендуются цитогенетические, генетические молекулярные и клинические анализы крови, миелограмма. Основными методами лечения являются трансплантация костного мозга, медицинское обеспечение кроветворения, переливание крови.
Анемия Фанкони
Дополнительные факты
Синонимами анемии Фанкони являются врожденная панмиелопатия Фанкони, наследственная панмиелопатия. Болезнь получила свое название от швейцарского педиатра Гвидо Фанкони, который в 1927 году описал врожденную апластическую патологию на основе симптомов трех братьев. Анемия Фанкони является редким генетическим заболеванием, наследуемым по аутосомно-рецессивному принципу. Эпидемиологические показатели невысоки: на 1 больного ребенка приходится 350 тысяч младенцев. Распространенность одинакова между представителями мужского и женского пола; оно выше в общинах с разрешенными близкими родственниками, например, среди некоторых южноафриканских народов.
Причины
Болезнь наследственная; это развивается, когда дефектный ген передается от родителей ребенку. Было идентифицировано 15 генов, мутации которых проявляются анемией Фанкони. Из них 14 расположены в аутосомах и являются рецессивными, 1 тип гена расположен в Х-хромосоме (связан с полом). Все эти гены отвечают за выработку определенного фермента, участвующего в репарации ДНК.
Аутосомно-рецессивное наследование подразумевает, что отец и мать должны нести патологическую генетическую информацию. Кроме того, они вообще здоровы. Вероятность рождения больного ребенка в такой паре составляет 25%. Генетическая панмиелопатия диагностируется у детей и взрослых, которые получают один и тот же измененный ген от каждого родителя. В очень редких случаях анемия вызвана передачей дефектной Х-хромосомы. Женщины могут нести мутации, болезнь проявляется только у мальчиков. Риск развития патологии у сына, если у матери мутированный ген, составляет 50%.
Патогенез
Обычно в клетках организма существуют специальные ферментативные системы, которые корректируют разрывы молекул ДНК, поврежденных при биосинтезе или воздействии химических, физических реагентов. При анемии Фанкони в кластере белков, ответственных за репарацию ДНК, обнаружен генетический дефект, что приводит к повышенной ломкости хромосом. В результате у пациентов развивается дисфункция костного мозга – рак и апластическая анемия. Онкологические заболевания чаще всего представлены острым миелоидным лейкозом – злокачественной опухолью костного мозга, которая вызывает накопление измененных лейкоцитов, которые ингибируют рост эритроцитов, тромбоцитов и нормальных лейкоцитов. При апластической анемии рост и созревание всех трех типов клеток крови быстро ингибируются дисплазией костного мозга.
Симптомы
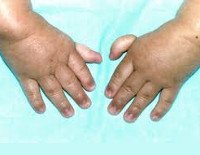
Более половины пациентов имеют врожденные аномалии внутренних органов и скелета. Деформации костей проявляются специфическим внешним видом: пациенты имеют низкорослый вид, уменьшенный размер головы, отсутствие или заметное укорочение большого пальца на руках, недоразвитие радиуса, врожденный вывих бедра и / или наличие шейного ребра, косолапость недоразвитый подбородок («птичье лицо»). Характерна гиперпигментация кожи в виде светлых и коричневатых пятен.
Неврологические расстройства представлены косоглазием, недоразвитием одного или двух глаз, опущением верхнего века, дрожанием глаз, глухотой, умственной отсталостью. Пациенты часто имеют незрелые половые органы, им не хватает одного или обоих яичек. Общие нарушения в структуре органов включают дефекты мочевыделительной системы: удвоение мочеточников или таза, подковообразные почки, почечные кисты, смещение наружного отверстия мочеиспускательного канала (гипоспадия). Врожденные пороки сердца включают атрезию трикуспидального клапана, дефект межпредсердной перегородки, митральный стеноз, дефект межжелудочковой перегородки. Пациенты страдают от почечной и сердечной недостаточности.
Основные симптомы связаны с постепенным увеличением нарушений костного мозга. Чаще всего они дебютируют в детстве (от 5 до 10 лет). Из-за уменьшения количества тромбоцитов усиливается кровотечение: кровь не сгущается в течение длительного времени, носовые кровотечения возникают легко, выделения во время менструации обильные, много «беспричинных» синяков на теле. Снижение количества эритроцитов проявляется анемией с характерной слабостью, быстрой утомляемостью, головокружением, обмороком, бледностью кожи, сердцебиением и одышкой. Дефицит лейкоцитов способствует ухудшению устойчивости к инфекциям. Впоследствии образуются лейкемия, миелодиспластический синдром и рак.
Вялость. Одышка.
Возможные осложнения
Частые инфекционные заболевания считаются наиболее распространенным осложнением. У пациентов развиваются ОРВИ, тонзиллит, ринит, бронхит, грипп, брюшной тиф и герпес. Рецидивирующий характер заболевания и его тяжелое течение приводят к разрушению органов, сопровождающемуся риском сепсиса. Еще одним осложнением наследственной анемии являются злокачественные новообразования – лейкемия, эпителиальные опухоли шеи и головы, половых органов. Рак у этих пациентов трудно поддается лечению из-за повышенной хрупкости и уменьшения восстановления ДНК. Это явление ограничивает применение лучевой терапии цитотоксическими препаратами. Нарушение свертываемости крови становится причиной большой потери крови.
Диагностика
Обследование пациентов проводится онкологами, гематологами, педиатрами и генетиками. Диагноз начинается с анализа анамнестических данных и жалоб. Врач выясняет, страдают ли эти родственники от этого наследственного заболевания, указывает время, в которое появляются первые признаки заболевания, ранние визиты к врачу. Во время обследования оценивается общее состояние пациента и выявляется наличие нарушений развития, гиперпигментированных пятен, кровотечений и кровоподтеков. В большинстве случаев нетрудно определить типичные деформации костей и недоразвитие глаз. Чтобы подтвердить диагноз и отличить анемию Фанкони от приобретенной анапластической анемии, проводится ряд лабораторных исследований:
• Клинический анализ крови. Изменения клеточного состава крови характерны. Тромбоцитопения и лейкопения диагностируются на ранних стадиях кроветворения, а панцитопения (резкое уменьшение объема эритроцитов, лейкоцитов и тромбоцитов) на поздних стадиях. Небольшой гемолиз возможен без гипербилирубинемии, но с ретикулоцитозом. Значение СОЭ возросло до 60-80 мм /.
• Цитогенетическое исследование клеток. Тест проводится с диэпоксибутаном, митомицином С, с указанием частоты и спектра хромосомных аберраций. В пользу генетической анемии параметры теста DEB считаются более 45%, пограничный уровень составляет 11-45% (процент клеток с хромосомными нарушениями).
• Молекулярно-генетический анализ клеток. Изучены гены, в которых мутации могут привести к развитию заболевания. В 60-70% случаев мутации обнаруживаются в паре генов FANCA, в 14% в аллеле FANCC и в 10% в генах FANCG. Уровень мутаций в других парах составляет 0,2-3%. Согласно исследованию, установлено увеличение количества фагоцитарных жиров в плазматических клетках и макрофагах. Содержание недифференцированных клеток находится в пределах нормы. Концентрация зародышевых клеток миелоцитов была снижена, а количество лимфоцитов увеличено.
Лечение
Основная терапия направлена на восстановление процесса кроветворения. Методы лечения подбираются индивидуально, в зависимости от тяжести заболевания, возраста пациента, наличия и тяжести врожденных дефектов. Кроме того, лечатся инфекции и онкопатологии и принимаются меры по реабилитации. Для устранения анемии используются следующие методы:
• BMT. Трансплантация костного мозга наиболее эффективна в отдаленном периоде, но имеет противопоказания, часто сопровождается развитием осложнений. Оптимальный возраст для операции – до десяти лет. Здоровые сестры и братья, которые соответствуют критериям отбора, могут быть донорами. Предварительная интенсивная терапия (кондиционирование) связана с риском токсического воздействия на органы. После трансплантации вероятность острого или хронического иммунного конфликта между донорскими и реципиентными клетками остается высокой.
• Медикаментозная стимуляция кроветворения. Если трансплантация невозможна, пациентам показано консервативное лечение, которое временно улучшает их состояние. Продукцию клеток крови стимулируют андрогены (мужские половые гормоны) и гематопоэтические факторы роста – эритропоэтин, фактор стволовых клеток, интерлейкины-1-12. Параллельно используются иммунодепрессанты. Медикаментозная терапия на протяжении многих лет поддерживала высокое качество жизни пациентов, но ее эффективность постепенно снижалась.
• Переливание компонентов крови. При тяжелых побочных эффектах или противопоказаниях к этиотропной терапии (трансплантация, стимуляция кроветворения) назначаются процедуры переливания крови. Промывают эритроциты, донорские эритроциты освобождаются от поверхностных белков, переливаются. При кровотечении и снижении уровня тромбоцитов пациентам вводят массу тромбоцитов.
Список литературы
1. Генетическая диагностика анемии Фанкони. Обзор литературы/ Панферова А. В. Тимофеева Н. М. Ольшанская Ю. В. // Онкогематология. – 2016.
2. Анемии (клиника, диагностика, лечение). Методическое пособие/ Филатов Л. Б. – 2006.
3. Свободнорадикальный статус клеток крови и костного мозга у детей с острыми лейкозами и анемией Фанкони: Автореферат диссертации/ Чивилева И. Ю. – 1996. V.
4. Анемия Фанкони: современная диагностика и терапия: Автореферат дисертации/ Кравченко Е. Г. – 2003.
Источник
Что такое анемия Фанкони?
Анемия Фанкони (сокр. АФ) — редкое генетическое заболевание в категории синдромов наследственной недостаточности костного мозга. Половина пациентов диагностируется до 10 лет, а около 10% заболевания диагностируются уже во взрослой жизни.
Больные пациенты страдают врожденными дефектами, такими как небольшой рост, ненормально большие пальцы и/или лучевые кости, пигментация кожи, маленькая голова, маленькие глаза, аномальные структуры почек, а также аномалии сердца и скелета.
Нарушение часто связано с прогрессирующим дефицитом всей продукции костного мозга клеток крови — эритроцитов, лейкоцитов и тромбоцитов. Пострадавшие люди имеют повышенный риск развития рака кроветворных клеток в костном мозге, называемого острым миелоидным лейкозом (ОМЛ), или опухолей головы, шеи, кожи, желудочно-кишечного тракта или половых путей.
Анемия Фанкони встречается одинаково у мужчин и женщин, и во всех этнических группах. Обычно он наследуется как аутосомно-рецессивное генетическое заболевание, но также сообщалось о Х-сцепленном наследовании.
Существует несколько подтипов АФ, которые являются результатом наследования двух генных мутаций в каждом из по меньшей мере 18 различных генов. Большинство подтипов имеют характерные симптомы и признаки. Анемия Фанкони — это не то же самое, что синдром Фанкони, редкое нарушение функции почек.
Признаки и симптомы
Симптомы анемии Фанкони варьируются от человека к человеку. Выявленные симптомы включают различные физические отклонения, недостаточность костного мозга и повышенный риск развития злокачественных новообразований. Физические отклонения обычно проявляются в раннем детстве, но в редких случаях диагнозы ставятся во взрослом возрасте. Проблемы с производством крови часто развиваются в возрасте 6-8 лет.
В большинстве случаев пораженный костный мозг встречается у большинства больных, хотя прогрессирование и возраст возникновения различаются. Пациенты, которые живут в зрелом возрасте, могут развить рак головы и шеи, гинекологический и/или желудочно-кишечный рак в гораздо более раннем возрасте, чем население в целом, независимо от того, были ли у них более ранние проблемы с кровью.
— Физические аномалии.
По крайней мере 60% людей, страдающих АФ, рождаются как минимум с одной физической аномалией. Аномалия могут включать любое из следующего:
- низкий рост;
- аномалии большого пальца и руки: лишние пальцы, деформация или отсутствие больших пальцев или не полностью развитый или отсутствие одной из костей предплечья;
- аномалии скелета бедер, позвоночника или ребер;
- структурные проблемы с почками;
- пигментация кожи;
- маленькая голова;
- маленькие, скрещенные или широко расставленные глаза;
- низкая масса тела при рождении;
- желудочно-кишечные проблемы;
- маленькие репродуктивные органы у мужчин;
- дефекты в тканях, разделяющих камеры сердца.
Люди с анемией могут испытывать:
- усталость;
- повышенную потребность во сне;
- слабость;
- головокружение;
- раздражительность;
- головные боли;
- бледный цвет кожи;
- затрудненное дыхание;
- сердечные симптомы.
Могут быть чрезмерные кровоподтеки после минимальной травмы и спонтанные кровотечения из слизистых оболочек, особенно десен и носа.
— Недостаточность костного мозга.
Костный мозг — губчатое вещество, находящийся в центре длинных костей тела. Костный мозг производит специализированные клетки (гемопоэтические стволовые клетки), которые растут и в конечном итоге развиваются в эритроциты (красные кровяные клетки), лейкоциты (белые кровяные клетки) и тромбоциты. Клетки попадают в кровоток, чтобы путешествовать по всему телу, выполняя свои специфические функции. Красные кровяные клетки доставляют кислород в организм, белые кровяные клетки помогают бороться с инфекциями, а тромбоциты позволяют организму образовывать сгустки, останавливая кровотечение.
Прогрессирующая недостаточность костного мозга обычно проявляется к 10 годам и обычно сопровождается низким уровнем тромбоцитов или низким уровнем лейкоцитов. К возрасту 40-50 лет предполагаемая частота недостаточности костного мозга как первого серьезного события составляет более 50%.
У пострадавших развивается низкий уровень всех клеточных элементов костного мозга — красных и белых кровяных клеток и тромбоцитов, что может привести к следующему:
- низкому уровню циркулирующих эритроцитов — анемия;
- низкому уровню лейкоцитов — лейкопения;
- низкому уровню нейтрофилов (тип лейкоцитов) — нейтропения;
- низкому уровню тромбоцитов — тромбоцитопения;
— Повышенный риск развития злокачественных новообразований.
Люди с АФ имеют более высокий риск развития определенных форм рака, включая острую миелоидную лейкемию и специфические солидные опухоли, чем население в целом.
Пострадавшие люди могут иметь чрезвычайно высокий риск развития рака, поражающего область головы и шеи, желудочно-кишечный тракт, пищевод или гинекологические области. Большинство из них представляют собой специфическую форму рака, известную как плоскоклеточный рак. У пациентов с АФ, у которых недостаточность костного мозга лечится мужскими гормонами (так называемыми «андрогенами»), повышен риск развития рака печени.
Примерно в 30 процентах случаев, связанных с раком, развитие злокачественной опухоли предшествует диагнозу АФ.
Причины анемии Фанкони
Хромосомы в клетках индивидуумов с анемией Фанкони не способны восстановить повреждение дезоксирибонуклеиновой кислоты (ДНК) и, таким образом, легко разрушаются и перестраиваются (нестабильность хромосом). ДНК является носителем генетического кода, а повреждение ДНК — обычное ежедневное явление. У большинства людей повреждение ДНК устраняется. Однако, у людей с АФ переломы и перестройки происходят чаще, и их тела медленны или не могут восстановить повреждения.
Мутации по крайней мере в 18 генах могут вызывать АФ. Белки, кодируемые этими генами, работают вместе по общему пути, называемому путем FA, который вступает в действие, когда происходит повреждение ДНК. Путь FA направляет определенные белки в область повреждения, так чтобы ДНК могло восстанавливаться и продолжать копироваться (реплицироваться). Восемь белков образуют комплекс, известный как комплекс ядра FA, который активирует два гена для образования белков, называемых FANCD2 и FANCI. Активация этих двух белков приводит белки репарации ДНК в область повреждения ДНК.
80-90% случаев заболевания происходят из-за мутаций в одном из трех генов, FANCA, FANCC и FANCG. Эти гены предоставляют инструкции для получения компонентов комплекса ядра FA. Мутации в любом из множества генов, связанных с основным комплексом FA, приведут к тому, что комплекс будет нефункциональным и нарушит весь путь FA. Нарушение этого пути приводит к накоплению повреждений ДНК, которые могут привести к аномальной гибели клеток или к их аномальному росту. Гибель клеток приводит к уменьшению количества клеток крови и физических нарушений, связанных с анемией Фанкони. Неконтролируемый рост клеток может привести к развитию острого миелоидного лейкоза или других видов рака.
Большинство случаев АФ наследуются по аутосомно-рецессивному типу. Рецессивные генетические нарушения возникают, когда человек наследует две копии ненормального гена по одному признаку, по одной от каждого родителя. Если человек наследует один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно не проявляет симптомов. Риск для двух родителей-носителей, которые оба передадут измененный ген и заразят ребенка, составляет 25% с каждой беременностью. Риск зачать ребенка, который является носителем как родители, составляет 50% с каждой беременностью. Вероятность для ребенка получить нормальные гены от обоих родителей составляет 25%. Риск одинаков для мужчин и женщин.
Родители, которые являются близкими родственниками (брат и сестра), имеют больше шансов, чем несвязанные родители, иметь один и тот же аномальный ген, что повышает риск рождения детей с рецессивным генетическим расстройством.
Мутации в следующих генах также вызывают АФ и наследуются по аутосомно-рецессивному типу: BRCA2, BRIP1, FANCB, FANCD2, FANCE, FANCF, FANCI, ERCC4, FANCL, FANCM, PALB2, RAD51C, SLX4 и UBE2T.
Ген FANCB расположен на Х-хромосоме и вызывает менее 1% всех случаев АФ. Этот ген наследуется как X-сцепленный рецессивный признак.
Х-связанные генетические расстройства — это состояния, вызванные ненормальным геном на Х-хромосоме и проявляющиеся в основном у мужчин. Женщины с измененным геном, присутствующим на одной из их Х-хромосом, являются носителями расстройства. Женщины-носители обычно не проявляют симптомов, поскольку у женщин есть две Х-хромосомы, и только одна несет измененный ген. У мужчин есть одна Х-хромосома, которая унаследована от матери, и если мужчина наследует Х-хромосому, которая содержит измененный ген, у него разовьется болезнь. Женщины-носители расстройства, связанного с Х, имеют 25% вероятность с каждой беременностью иметь дочь-носительницу, подобную себе, 25% вероятность иметь дочь, не являющуюся носителем, 25% вероятность иметь сына, пораженного этой болезнью, и 25% вероятность иметь незатронутого сына. Если мужчина с Х-сцепленным расстройством способен размножаться, он передаст измененный ген всем своим дочерям, которые будут носительницами. Мужчина не может передать свой X-связанный ген своим сыновьям, поскольку мужчины всегда передают свою Y-хромосому вместо своей X-хромосомы потомству мужского пола.
Мутации в гене RAD51 вызывают аутосомно-доминантный АФ. Доминантные генетические расстройства возникают, когда только одна копия ненормального гена необходима для того, чтобы вызвать конкретное заболевание. Аномальный ген может быть унаследован от любого из родителей или может быть результатом новой мутации (изменения гена) у пострадавшего человека. Риск передачи ненормального гена от пострадавшего родителя потомству составляет 50% для каждой беременности. Риск одинаков для мужчин и женщин. На сегодняшний день все больные люди с АФ вследствие мутации гена RAD51 имеют спонтанную (de novo) генетическую мутацию, которая происходит в яйцеклетке или сперматозоиде. В таких ситуациях расстройство не наследуется от родителей.
Затронутые группы населения
По оценкам, уровень заболеваемости анемией Фанкони составляет около 1 на 136 000 рождений. Заболевание более распространено среди евреев-ашкенази, цыган в Испании и чернокожих южноафриканцев.
Диагностика
Диагноз АФ ставится на основании тщательной клинической оценки, подробного анамнеза пациента, определения характерных признаков и различных специализированных тестов.
Окончательным тестом на АФ в настоящее время является тест на разрыв хромосом: некоторые клетки крови пациента обрабатывают в пробирке химическим веществом, которое сшивает ДНК. Нормальные клетки способны исправить большинство повреждений и не подвергаются серьезному воздействию, тогда как при заболевании клетки демонстрируют заметное разрушение хромосом. Для этого теста обычно используются два химических вещества: DEB (диэпоксибутан) и MMC (митомицин C). Эти тесты могут быть выполнены пренатально на клетках из ворсин хориона или из околоплодных вод.
Анализы крови могут быть выполнены, чтобы определить уровни красных и белых клеток крови и тромбоцитов. Рентгенологическое исследование может выявить наличие и степень пороков развития скелета и внутренних структурных аномалий.
Многие случаи анемии Фанкони вообще не диагностируются или не диагностируются своевременно. АФ следует заподозрить и проверить на наличие у любого ребенка, родившегося с аномалиями большого пальца и руки, описанными ранее. Любой, у кого развивается апластическая анемия в любом возрасте, должен пройти тест на АФ, даже если других дефектов нет. Любой пациент, у которого в раннем возрасте развивается плоскоклеточный рак головы и шеи, желудочно-кишечного тракта или гинекологической системы с или без употребления табака или алкоголя, должен пройти обследование на АФ. Многие пациенты с АФ не показывают никаких других отклонений. Необходимо провести тест на АФ, прежде чем рассматривать трансплантацию стволовых клеток на предмет апластической анемии или лечения рака, поскольку стандартные протоколы химиотерапии и облучения могут оказаться токсичными для пациентов с заболеванием.
Молекулярно-генетическое тестирование доступно для всех 18 генов, связанных с АФ. Тестирование комплементации обычно проводится первым, чтобы определить, какой ген мутирует. Затем можно провести анализ последовательности соответствующего гена, чтобы определить конкретную мутацию в этом гене. Если мутация не идентифицирована, клинически доступен анализ делеции/дупликации генов.
Целевой мутационный анализ доступен для обычной ашкеназской еврейской мутации FANCC.
— Клиническое исследование.
Чтобы определить степень заболевания у индивидуума с диагнозом АФ, при необходимости рекомендуется следующие исследования:
- Ультразвуковое исследование почек и мочевыводящих путей.
- Формальный тест слуха.
- Оценка развития (особенно важно для малышей и детей школьного возраста).
- Обращение к офтальмологу, отоларингологу, эндокринологу, хирургу руки, гинекологу (для женщин, как указано), гастроэнтерологу, урологу, дерматологу, ЛОР-хирургу, генетическому консультанту.
- Оценка гематологом, включая полный анализ крови, гемоглобина плода и аспирата костного мозга для морфологии клеток и исследования хромосом (цитогенетика), а также биопсию для определения клеточности.
- HLA-типирование отдельных лиц, братьев и сестер и родителей для рассмотрения трансплантации гемопоэтических стволовых клеток.
- Полное типирование крови.
- Химия крови (оценка состояния печени, почек, щитовидной железы, липидов и железа).
Лечение анемии Фанкони
Лечение анемии Фанкони направлено на конкретные симптомы, которые проявляются у каждого человека. Лечение может потребовать скоординированных усилий команды специалистов. Педиатрам, хирургам, кардиологам, почечным специалистам (нефрологам), урологам, гастроэнтерологам, специалистам, которые оценивают и лечат проблемы со слухом (аудиологам и отоларингологам), глазным специалистам и другим специалистам здравоохранения, возможно, потребуется систематически и всесторонне планировать лечение пострадавшего.
Рекомендации по лечению были согласованы на консенсусной конференции 2014 года (https://www.nhlbi.nih.gov/health/health-topics/topics/fanconi/).
- Введение андрогенов (мужской гормон): андрогены улучшают показатели крови примерно у 50% людей с АФ. Самый ранний ответ наблюдается в эритроцитах, при этом увеличение гемоглобина обычно происходит в течение первого или двух месяцев лечения. Ответы на количество белых клеток и количество тромбоцитов являются переменными. Реакции тромбоцитов, как правило, неполные и не могут быть замечены до нескольких месяцев терапии. Улучшение, как правило, самое большое для количества эритроцитов. Сопротивление терапии может развиваться со временем.
- Гематопоэтические факторы роста: гранулоцитарный колониестимулирующий фактор (G-CSF) может улучшать количество нейтрофилов у некоторых людей. Обычно используется только для поддержки при интеркуррентных заболеваниях.
- Гематопоэтическая трансплантация стволовых клеток (HSCT): единственная лечебная терапия для гематологических проявлений АФ. Донорские стволовые клетки могут быть получены из костного мозга, периферической крови или пуповинной крови.
- Лечение рака: Лечение злокачественных новообразований является сложной задачей в связи с повышенной токсичностью, связанной с химиотерапией и облучением при анемии Фанкони. Следует проявлять осторожность в центрах, имеющих опыт лечения пациентов с АФ.
Хирургическое вмешательство может быть необходимо для коррекции пороков развития скелета, таких как поражения больших пальцев рук и костей предплечья, пороки сердца и желудочно-кишечные нарушения, такие как трахеопищеводный свищ или атрезия пищевода, а также анальная атрезия.
Некоторые химические вещества могут увеличивать риск хромосомных нарушений у людей с АФ, и их следует избегать, когда это возможно. Эти химические вещества включают табачный дым, формальдегид, гербициды и органические растворители, такие как бензин или разбавитель краски.
Рекомендуется генетическое консультирование для пострадавших людей и их семей.
Источник