Анемия из за хромосом

Анемия Фанкони (АФ), или панцитопения Фанкони, является синдромом нестабильности генома. Это редкое наследственное аутосомно-рецессивное заболевание с вариабельной пенетрантностью и генетической гетерогенностью. АФ была впервые описана в 1927 г. швейцарским педиатром Гвидо Фанкони, который сообщил о 3 братьях с панцитопенией и пороками физического развития. Термин «анемия Фанкони» был предложен Негели в 1931 г. для обозначения комбинации семейной анемии Фанкони и врожденных физических пороков. По настоящее время описано чуть более 2000 случаев АФ. Частота гетерозиготного носительства существенно различается в разных популяциях. Традиционно указывалась цифра 1:300, по последним данным североамериканского регистра, она составляет 1:181, в Израиле — 1:93. Следует заметить, что около 6 % больных не имеют никаких аномалий развития. Некоторые мутации этнически закреплены, в основе их распространения лежит «эффект основателя» — потеря генетической вариабельности в популяциях, основанных малым количеством предков, что свойственно для относительно небольших популяций. Эти мутации встречаются у евреев-ашкенази, испанских цыган, голландцев, выходцев с Канарских островов, у жителей Южной Африки и Кореи. В некоторых из этих популяций частота носительства этнически закрепленных мутаций довольно высока и оценена приблизительно как 1:100. Частота встречаемости тех или иных мутаций в РФ не изучена.
Этиология и патогенез
Нарушение структуры ДНК является результатом воздействия как внутренних (депуринизация, дезаминирование, воздействие эндогенных альдегидов, особенно формальдегида, и активных форм кислорода), так и внешних факторов (ионизирующее и УФ облучение, химические мутагены). При воздействии этих факторов происходят реакции алкилирования, окисления, восстановления, связывания с формальдегидными группами азотистых оснований. В итоге возникают изменения одного или нескольких оснований, вставки и делеции, образование тиминовых димеров, одно- и двухцепочечные разрывы ДНК, образование сшивок между основаниями одной цепи или комплементарными цепями ДНК, между ДНК и белковыми молекулами. Несмотря на это, в целом геном остается свободным от «ошибок», так как клетка имеет механизмы детекции и репарации поврежденной ДНК. Поврежденное основание может быть восстановлено непосредственно его заменой или обратной химической реакцией (direct repair), в других случаях необходимы более сложные процессы, обеспечивающие удаление поврежденного участка ДНК и достраивание правильной последовательности с использованием комплементарной цепи, редко — гомологичной хромосомы. Такая репарация ДНК — сложный многоступенчатый процесс взаимодействия нескольких каскадных путей. Процесс репарации происходит на разных этапах клеточного цикла. При АФ нарушается способность клетки исправлять определенный тип повреждений ДНК — поперечные межхроматидные сшивки (DNA interstrand crosslink), которые препятствуют работе репликационной вилки. Поперечные межхроматидные сшивки формируются как под воздействием продуктов естественного метаболизма клетки (в первую очередь эндогенных альдегидов, но также и активных форм кислорода), так и под воздействием химических веществ, в частности химиотерапевтических препаратов. К такому типу химических веществ относятся алкилирующие соединения, имеющие в своем составе две активные алкильные группы, обеспечивающие им активное связывание с определенными основаниями: цисплатин, митомицин С, азотистый иприт, псорален, диэпоксибутан, мелфалан, циклофосфамид, мустарген, стрептозоцин. Протеины, функция которых нарушается при АФ, задействованы во всех этапах репарации межхроматидной поперечной сшивки. Этот сложный многоступенчатый процесс получил название FA-pathway, а протеины, задействованные в нем, — АФ-протеины. Ключевую роль в этом процессе играет моноубиквитинирование гена FANCD2, который координирует процессы вырезки поврежденных нуклеотидов, прямое достраивание поврежденного участка и гомологичную рекомбинацию. При АФ клетка не способна адекватно исправлять повреждения ДНК, накопление поломок которой может приводить к костномозговой недостаточности, аномалиям физического развития и предрасположенности к развитию опухолей.
Спектр мутаций, которые приводят к АФ:
1) FANCA.
Мутации в этом гене — самые распространенные и встречаются в 60–70 % случаев АФ. Известно более 100 мутаций, из которых около трети приходится на точечные, еще треть представляют собой микроделеции, и около 40 % представлены крупными делециями. Описаны также и малые дупликации. По действию мутации в гене FANCA могут быть гипоморфными, т. е. приводить к частичной потере функции белка; они характеризуются более мягкими клиническими проявлениями, однако большая часть мутаций вызывает полную потерю функции. Ряд мутаций в гене FANCA имеет повышенную частоту распространения. Так, микроделеция в 38-м экзоне с.3788_3790delTCT — самая распространенная мутация при АФ в мире (20,7 % всех аллелей с мутацией). При этом она встречается у 80 % пациентов с АФ с Канарских островов, где частота встречаемости АФ достигает 1:16000 новорожденных. Кроме того, эта мутация встречается в 51 % случаев АФ в Бразилии. Для подтверждения «эффекта основателя» для данной мутации был проведен анализ гаплотипа пациентов путем изучения вариабельных тандемных повторов и однонуклеотидных полиморфизмов гена FANCA у 28 пациентов с мутацией с.3788_3790delTCT из различных частей света. Все, за исключением одного пациента, имели общего предка. По всей видимости, Канарские острова послужили местом происхождения и распространения заболевания из Европы в Америку, так как несколько веков назад практически все суда из Испании в Америку шли через Канарские острова. Однако, учитывая, что эта мутация тем не менее составляет 2–5 % всех FANCA-мутаций, существует также и мнение, что она связана с явлением существования определенных участков генома с повышенной мутационной способностью, так называемых hotspot. Другой пример «эффекта основателя» — мутация с.295С>Т, которую выявляют почти во всех случаях АФ у испанских цыган. При этом носительство этой мутации среди испанских цыган определено как 1:67. Функционально и фенотипически варианты мутаций в гене FANCA проявляются одинаково.
2) FANCC.
Мутации в гене FANCC встречаются в 10–15 % случаев АФ, почти 90 % случаев представлено двумя мутациями — с.711+4А>Т и delG332. Самая частая мутация в гене FANCC — с. 711+4А>Т, ее выявляют в гомозиготном состоянии в 80 % случаев АФ у евреев-ашкенази. Частота гетерозиготного носительства этой мутации среди евреев-ашкенази достигает 40 %. При этом встречаются и спорадические случаи. Генотип FANCC с.711+4А>Т также распространен среди больных АФ в Японии. В Нидерландах более чем 50 % случаев АФ — это гомозиготные носители мутации с.67delG (также известна как delG332), приводящей к сдвигу рамки считывания в гене FANCC.
3) FANCG.
FANCG задействован в 10 % случаев АФ. Встречаются практически все типы мутаций, за исключением крупных делеций. Клинически характеризуются более частым и быстрым развитием миелодисплазии или лейкоза. Встречаются как спорадические случаи, так и этнически ассоциированный вариант — около 80 % случаев АФ у чернокожих южноафриканцев (Bantu-speakers) (Южная Африка, Свазиленд, Малави и Мозамбик) имеют мутацию 637_643delTACCGCC. Заболевание характеризуется частыми нарушениями пигментации кожи, слабовыраженными аномалиями развития и сравнительно поздними, но тяжелыми гематологическими проявлениями, обусловленными в том числе и поздним обращением к врачу.
Развитие костномозговой недостаточности связывают с повышенным апоптозом гемопоэтических клеток, однако истинные патогенетические механизмы костномозговой недостаточности при АФ мало изучены из-за сложности получения адекватной биологической модели развития заболевания. Последние исследования на ксенографтных моделях и in vitro показали, что в ответ на накопление нерепарированных повреждений ДНК происходит активация р53 проапоптотического пути и запуск поздней р21(Cdkn1a)-зависимой блокировки клеточного цикла в фазе G0/G1 с последующей элиминацией ранних гемопоэтических предшественников из костного мозга. Этот механизм запускается в пренатальном периоде на этапе формирования пула стволовых клеток и ранних клеток-предшественников гемопоэза, что приводит к значительному снижению их количества. Накопление дефектов ДНК после рождения в результате различных физико-химических воздействий усугубляет нарушение гемопоэза. Кроме выраженного апоптоза ранних клеток-предшественников происходит нарушение базовых свойств стволовых кроветворных клеток — способности к самоподдержанию собственной популяции, пролиферации и дифференцировке в различные линии гемопоэза. Генетическая нестабильность при АФ реализуется в повышенной частоте развития ряда опухолей, наиболее частые — ОМЛ и плоскоклеточный рак кожи головы и шеи, слизистых оболочек рта и мочеполового тракта — то есть тканей, характеризующихся высокой пролиферативной активностью.
Клиническая картина
Признаки и симптомы, а также частота их встречаемости указаны в таблице 1.
Таблица 1 | Признаки и симптомы анемии Фанкони
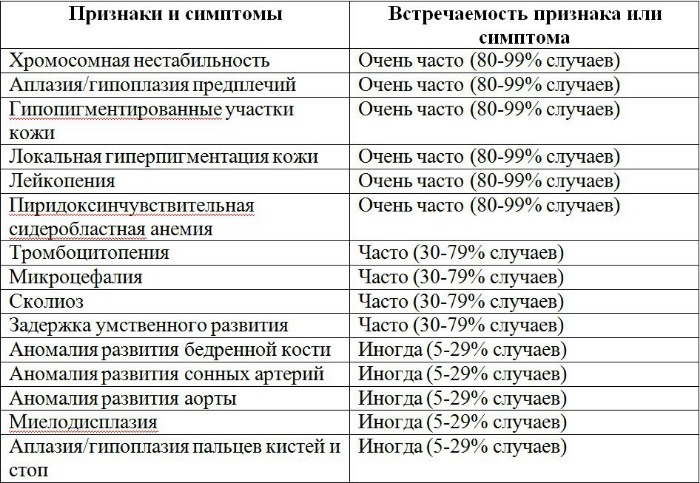
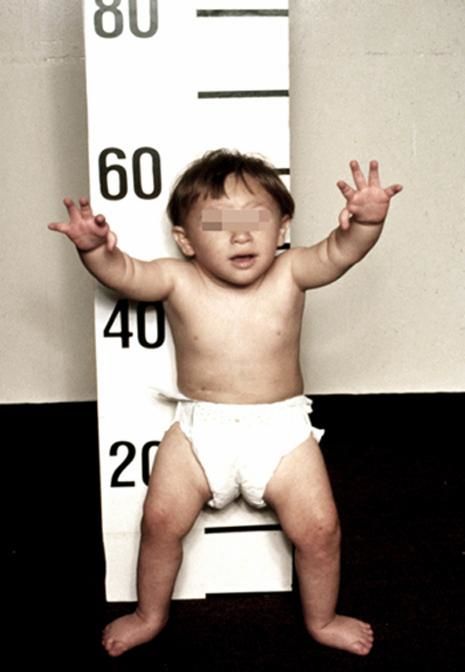
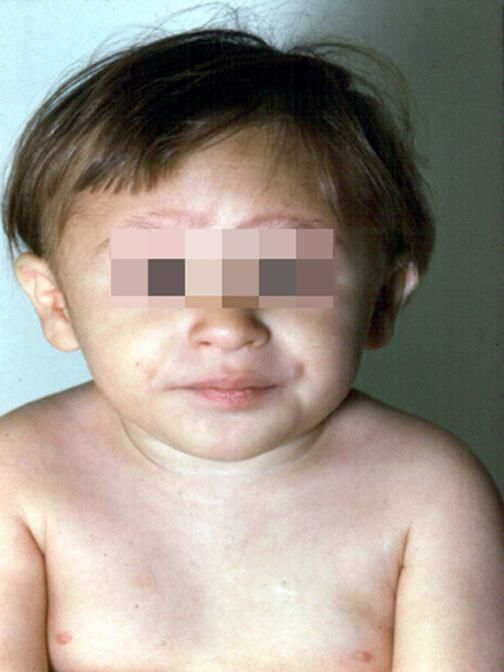
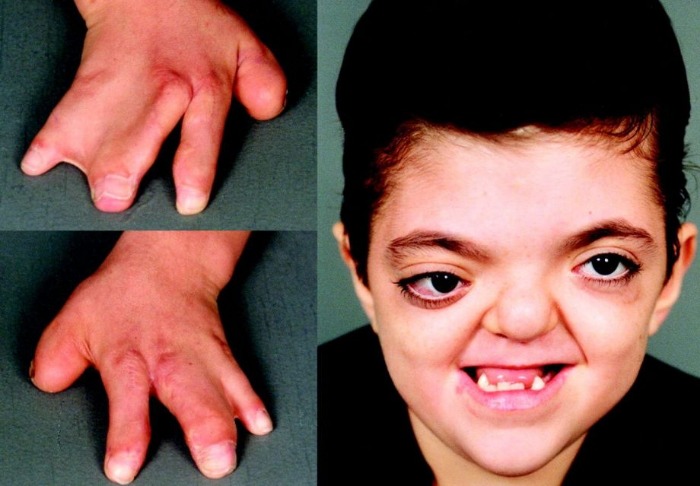
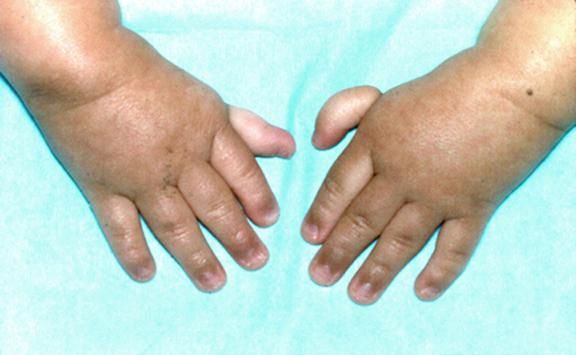
Рисунки 1–5 | Пороки развития, наблюдаемые при анемии Фанкони.
Диагностика
Помимо тщательного физикального осмотра, сбора семейного анамнеза, общего анализа крови и пункции костного мозга для более точной диагностики используются такие методы лабораторной диагностики, как тест на ломкость хромосом, метод MLPA, высокопроизводительное секвенирование и секвенирование по Сэнгеру.
1) Тест на ломкость хромосом.
«Золотым стандартом» скрининга для выявления АФ был и остается тест с диэпоксибутаном (1,3-butadienediepoxide) и его вариант с митомицином С (MMC). Еще в начале изучения АФ было отмечено, что фибробласты и лимфоциты больных АФ в культуре клеток демонстрируют спонтанную повышенную ломкость хромосом. Значительные различия в уровне спонтанных аберраций у больных (вплоть до отсутствия таковых) требовали унифицированного и точного метода детекции. Позже была показана повышенная чувствительность клеток больных АФ к действию алкилирующих агентов, вызывающих поперечные сшивки между нуклеотидами, что препятствует образованию нормальной репликативной вилки для запуска процесса репарации ДНК, позже получивших общее название interstrand cross-link agent. На основании этого был предложен цитогенетический метод диагностики АФ: после обработки лимфоцитов или фибробластов алкилирующим веществом (в нелетальной для клеток концентрации) определяют частоту и спектр спонтанных и индуцированных in vitro хромосомных аберраций. Обычно ставится несколько параллельных клеточных культур, стимулированных фитогемагглютинином лимфоцитов периферической крови: без добавления алкилирующего агента (для определения спонтанного уровня аберраций) и с его добавлением в разной концентрации. Затем в метафазных пластинках подсчитывают число хромосомных разрывов. Для АФ характерны разрывы хромосом с образованием радиальных фигур, фрагментов, хромосомных и хроматидных разрывов, а также три- и тетрарадикалов. При анализе результатов учитывают число разрывов по отношению к числу проанализированных метафаз, процент клеток с разрывами и ряд других показателей. Необходимо отметить, что значения, при которых тест на ломкость хромосом считается положительным, в различных лабораториях варьируются. Тест не имеет 100 % специфичности. Положительный тест на ломкость хромосом бывает у пациентов с синдромом Ниймеген (мутации в гене NBS1), синдромом Робертса (мутации в гене ESCO2), Warsaw breakage syndrome (мутации в гене DDX11), синдромом Блюма (ген BLM), врожденном дискератозе и некоторых других синдромах. В то же время АФ-подобные синдромы могут оказаться действительно АФ с соответствующим генетическим дефектом.
2) Секвенирование по Сэнгеру долгие годы было основным методом определения мутаций при АФ. Учитывая значительные размеры генов, секвенирование каждого из них представляет собой довольно трудоемкий и дорогостоящий процесс. Его обычно проводили после анализа на группу комплементации, предварительно определив вероятный ген. Анализ нуклеотидной последовательности для всех известных АФ-генов затруднен количеством возможных мутаций в каждом, их разнообразием, в том числе в виде крупных инсерций или делеций (indel-мутации). Их длина может варьировать от одного до нескольких сотен и даже тысяч нуклеотидных оснований, что подразумевает использование совершенно разных методов молекулярно-генетического исследования.
3) Метод MLPA (мультиплексная амплификация лигазносвязанных проб) предназначен для определения делеций и амплификаций определенных последовательностей гена длиной до нескольких десятков нуклеотидов. Одновременно может быть исследовано до 60 таких последовательностей, что позволяет выявить как сравнительно небольшие делеции, так и делеции отдельных экзонов и целого гена. Метод MLPA используют для инициального скрининга делеции в гене FANCA, параллельно ген FANCA полностью секвенируют. Если пациент мужского пола, его исследуют на наличие делеций ген FANCB. Выявленные делеции желательно подтвердить другим методом. При этом методы, которые могут быть использованы, требуют индивидуальной разработки в каждом конкретном случае: количественная ПЦР, ПЦР длинных фрагментов (long range PCR) и хромосомный микроматричный анализ.
4) Высокопроизводительное секвенирование.
Метод, который позволяет одномоментно анализировать от нескольких генов до полного генома, является наиболее подходящим для определения мутаций при АФ. Возможны несколько вариантов теста. Первый — секвенирование экзома, позволяет получить максимальный объем информации. Второй — секвенирование ограниченного числа интересующих и уже описанных в литературе генов (таргетное ресеквенирование), список которых можно дополнять или моделировать в соответствии с потребностями исследования. Современные коммерческие панели генов, как правило, помимо генов АФ включают большое число генов, ответственных за развитие других врожденных синдромов, в том числе и АФ-подобных. Внедрение в практику высокопроизводительного секвенирования позволяет избежать последовательного трудоемкого исследования каждого из известных генов методом секвенирования по Сэнгеру, однако пока не позволяет с должной уверенностью выявить крупные делеции и дупликации. Найденные мутации требуют подтверждения одним из других подходящих методов. При обнаружении новых мутаций необходимо подтверждение их патогенности в функциональном тесте и др.
Пренатальная и преимплантационная диагностика должна проводиться в первую очередь в семьях, где ранее были установлены патогенетические мутации. В этом случае проводится целенаправленный поиск известной мутации. Материалом для диагностики служат клетки плода, получаемые путем биопсии ворсин хориона на 10–12 неделе беременности. Следует помнить, что генетический анализ занимает не менее 2–3 недель. Если нет возможности провести молекулярно-генетическое исследование, возможно выполнение теста на ломкость хромосом клеток ворсин хориона на 10–12 неделе беременности либо при амниоцентезе на 15–18 неделе. Однако молекулярно-генетическое исследование предпочтительнее.
Лечение
Основной метод лечения АФ — аллогенная трансплантация гемопоэтических стволовых клеток.
Из медикаментозного лечения в настоящий момент применяют андрогены. Препараты этой группы (оксиметалон, метандростенолон) позволяют достичь гематологического ответа примерно у 50 % больных. Также применение андрогенов значимо увеличивает продолжительность жизни у ответивших на лечение пациентов: медиана продолжительности жизни составляет 9 лет после установления диагноза против 2,5 лет соответственно для тех пациентов, у которых лечение андрогенами не было эффективным.
При возникновении сопутствующих заболеваний, например, инфекций, обосновано применение гранулоцитарного колониестимулирующего фактора, который способен временно увеличить количество нейтрофилов и облегчить протекание заболевания.
В 2015 году коллективу ученых удалось успешно применить технологию Crispr/Cas9 для редактирования мутации с.67delG гена FANCC.
Источники:
- https://www.bloodjournal.org/content/126/23/3622?sso-checked=true
- https://www.ncbi.nlm.nih.gov/books/NBK1401/
- https://rarediseases.info.nih.gov/diseases/6425/fanconi-anemia
- https://rarediseases.org/rare-diseases/fanconi-anemia/
- https://cyberleninka.ru/article/v/geneticheskaya-diagnostika-anemii-fankoni-obzor-literatury
Нашли опечатку? Выделите фрагмент и нажмите Ctrl+Enter.
Источник
Что такое анемия Фанкони?
Анемия Фанкони (сокр. АФ) — редкое генетическое заболевание в категории синдромов наследственной недостаточности костного мозга. Половина пациентов диагностируется до 10 лет, а около 10% заболевания диагностируются уже во взрослой жизни.
Больные пациенты страдают врожденными дефектами, такими как небольшой рост, ненормально большие пальцы и/или лучевые кости, пигментация кожи, маленькая голова, маленькие глаза, аномальные структуры почек, а также аномалии сердца и скелета.
Нарушение часто связано с прогрессирующим дефицитом всей продукции костного мозга клеток крови — эритроцитов, лейкоцитов и тромбоцитов. Пострадавшие люди имеют повышенный риск развития рака кроветворных клеток в костном мозге, называемого острым миелоидным лейкозом (ОМЛ), или опухолей головы, шеи, кожи, желудочно-кишечного тракта или половых путей.
Анемия Фанкони встречается одинаково у мужчин и женщин, и во всех этнических группах. Обычно он наследуется как аутосомно-рецессивное генетическое заболевание, но также сообщалось о Х-сцепленном наследовании.
Существует несколько подтипов АФ, которые являются результатом наследования двух генных мутаций в каждом из по меньшей мере 18 различных генов. Большинство подтипов имеют характерные симптомы и признаки. Анемия Фанкони — это не то же самое, что синдром Фанкони, редкое нарушение функции почек.
Признаки и симптомы
Симптомы анемии Фанкони варьируются от человека к человеку. Выявленные симптомы включают различные физические отклонения, недостаточность костного мозга и повышенный риск развития злокачественных новообразований. Физические отклонения обычно проявляются в раннем детстве, но в редких случаях диагнозы ставятся во взрослом возрасте. Проблемы с производством крови часто развиваются в возрасте 6-8 лет.
В большинстве случаев пораженный костный мозг встречается у большинства больных, хотя прогрессирование и возраст возникновения различаются. Пациенты, которые живут в зрелом возрасте, могут развить рак головы и шеи, гинекологический и/или желудочно-кишечный рак в гораздо более раннем возрасте, чем население в целом, независимо от того, были ли у них более ранние проблемы с кровью.
— Физические аномалии.
По крайней мере 60% людей, страдающих АФ, рождаются как минимум с одной физической аномалией. Аномалия могут включать любое из следующего:
- низкий рост;
- аномалии большого пальца и руки: лишние пальцы, деформация или отсутствие больших пальцев или не полностью развитый или отсутствие одной из костей предплечья;
- аномалии скелета бедер, позвоночника или ребер;
- структурные проблемы с почками;
- пигментация кожи;
- маленькая голова;
- маленькие, скрещенные или широко расставленные глаза;
- низкая масса тела при рождении;
- желудочно-кишечные проблемы;
- маленькие репродуктивные органы у мужчин;
- дефекты в тканях, разделяющих камеры сердца.
Люди с анемией могут испытывать:
- усталость;
- повышенную потребность во сне;
- слабость;
- головокружение;
- раздражительность;
- головные боли;
- бледный цвет кожи;
- затрудненное дыхание;
- сердечные симптомы.
Могут быть чрезмерные кровоподтеки после минимальной травмы и спонтанные кровотечения из слизистых оболочек, особенно десен и носа.
— Недостаточность костного мозга.
Костный мозг — губчатое вещество, находящийся в центре длинных костей тела. Костный мозг производит специализированные клетки (гемопоэтические стволовые клетки), которые растут и в конечном итоге развиваются в эритроциты (красные кровяные клетки), лейкоциты (белые кровяные клетки) и тромбоциты. Клетки попадают в кровоток, чтобы путешествовать по всему телу, выполняя свои специфические функции. Красные кровяные клетки доставляют кислород в организм, белые кровяные клетки помогают бороться с инфекциями, а тромбоциты позволяют организму образовывать сгустки, останавливая кровотечение.
Прогрессирующая недостаточность костного мозга обычно проявляется к 10 годам и обычно сопровождается низким уровнем тромбоцитов или низким уровнем лейкоцитов. К возрасту 40-50 лет предполагаемая частота недостаточности костного мозга как первого серьезного события составляет более 50%.
У пострадавших развивается низкий уровень всех клеточных элементов костного мозга — красных и белых кровяных клеток и тромбоцитов, что может привести к следующему:
- низкому уровню циркулирующих эритроцитов — анемия;
- низкому уровню лейкоцитов — лейкопения;
- низкому уровню нейтрофилов (тип лейкоцитов) — нейтропения;
- низкому уровню тромбоцитов — тромбоцитопения;
— Повышенный риск развития злокачественных новообразований.
Люди с АФ имеют более высокий риск развития определенных форм рака, включая острую миелоидную лейкемию и специфические солидные опухоли, чем население в целом.
Пострадавшие люди могут иметь чрезвычайно высокий риск развития рака, поражающего область головы и шеи, желудочно-кишечный тракт, пищевод или гинекологические области. Большинство из них представляют собой специфическую форму рака, известную как плоскоклеточный рак. У пациентов с АФ, у которых недостаточность костного мозга лечится мужскими гормонами (так называемыми «андрогенами»), повышен риск развития рака печени.
Примерно в 30 процентах случаев, связанных с раком, развитие злокачественной опухоли предшествует диагнозу АФ.
Причины анемии Фанкони
Хромосомы в клетках индивидуумов с анемией Фанкони не способны восстановить повреждение дезоксирибонуклеиновой кислоты (ДНК) и, таким образом, легко разрушаются и перестраиваются (нестабильность хромосом). ДНК является носителем генетического кода, а повреждение ДНК — обычное ежедневное явление. У большинства людей повреждение ДНК устраняется. Однако, у людей с АФ переломы и перестройки происходят чаще, и их тела медленны или не могут восстановить повреждения.
Мутации по крайней мере в 18 генах могут вызывать АФ. Белки, кодируемые этими генами, работают вместе по общему пути, называемому путем FA, который вступает в действие, когда происходит повреждение ДНК. Путь FA направляет определенные белки в область повреждения, так чтобы ДНК могло восстанавливаться и продолжать копироваться (реплицироваться). Восемь белков образуют комплекс, известный как комплекс ядра FA, который активирует два гена для образования белков, называемых FANCD2 и FANCI. Активация этих двух белков приводит белки репарации ДНК в область повреждения ДНК.
80-90% случаев заболевания происходят из-за мутаций в одном из трех генов, FANCA, FANCC и FANCG. Эти гены предоставляют инструкции для получения компонентов комплекса ядра FA. Мутации в любом из множества генов, связанных с основным комплексом FA, приведут к тому, что комплекс будет нефункциональным и нарушит весь путь FA. Нарушение этого пути приводит к накоплению повреждений ДНК, которые могут привести к аномальной гибели клеток или к их аномальному росту. Гибель клеток приводит к уменьшению количества клеток крови и физических нарушений, связанных с анемией Фанкони. Неконтролируемый рост клеток может привести к развитию острого миелоидного лейкоза или других видов рака.
Большинство случаев АФ наследуются по аутосомно-рецессивному типу. Рецессивные генетические нарушения возникают, когда человек наследует две копии ненормального гена по одному признаку, по одной от каждого родителя. Если человек наследует один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно не проявляет симптомов. Риск для двух родителей-носителей, которые оба передадут измененный ген и заразят ребенка, составляет 25% с каждой беременностью. Риск зачать ребенка, который является носителем как родители, составляет 50% с каждой беременностью. Вероятность для ребенка получить нормальные гены от обоих родителей составляет 25%. Риск одинаков для мужчин и женщин.
Родители, которые являются близкими родственниками (брат и сестра), имеют больше шансов, чем несвязанные родители, иметь один и тот же аномальный ген, что повышает риск рождения детей с рецессивным генетическим расстройством.
Мутации в следующих генах также вызывают АФ и наследуются по аутосомно-рецессивному типу: BRCA2, BRIP1, FANCB, FANCD2, FANCE, FANCF, FANCI, ERCC4, FANCL, FANCM, PALB2, RAD51C, SLX4 и UBE2T.
Ген FANCB расположен на Х-хромосоме и вызывает менее 1% всех случаев АФ. Этот ген наследуется как X-сцепленный рецессивный признак.
Х-связанные генетические расстройства — это состояния, вызванные ненормальным геном на Х-хромосоме и проявляющиеся в основном у мужчин. Женщины с измененным геном, присутствующим на одной из их Х-хромосом, являются носителями расстройства. Женщины-носители обычно не проявляют симптомов, поскольку у женщин есть две Х-хромосомы, и только одна несет измененный ген. У мужчин есть одна Х-хромосома, которая унаследована от матери, и если мужчина наследует Х-хромосому, которая содержит измененный ген, у него разовьется болезнь. Женщины-носители расстройства, связанного с Х, имеют 25% вероятность с каждой беременностью иметь дочь-носительницу, подобную себе, 25% вероятность иметь дочь, не являющуюся носителем, 25% вероятность иметь сына, пораженного этой болезнью, и 25% вероятность иметь незатронутого сына. Если мужчина с Х-сцепленным расстройством способен размножаться, он передаст измененный ген всем своим дочерям, которые будут носительницами. Мужчина не может передать свой X-связанный ген своим сыновьям, поскольку мужчины всегда передают свою Y-хромосому вместо своей X-хромосомы потомству мужского пола.
Мутации в гене RAD51 вызывают аутосомно-доминантный АФ. Доминантные генетические расстройства возникают, когда только одна копия ненормального гена необходима для того, чтобы вызвать конкретное заболевание. Аномальный ген может быть унаследован от любого из родителей или может быть результатом новой мутации (изменения гена) у пострадавшего человека. Риск передачи ненормального гена от пострадавшего родителя потомству составляет 50% для каждой беременности. Риск одинаков для мужчин и женщин. На сегодняшний день все больные люди с АФ вследствие мутации гена RAD51 имеют спонтанную (de novo) генетическую мутацию, которая происходит в яйцеклетке или сперматозоиде. В таких ситуациях расстройство не наследуется от родителей.
Затронутые группы населения
По оценкам, уровень заболеваемости анемией Фанкони составляет около 1 на 136 000 рождений. Заболевание более распространено среди евреев-ашкенази, цыган в Испании и чернокожих южноафриканцев.
Диагностика
Диагноз АФ ставится на основании тщательной клинической оценки, подробного анамнеза пациента, определения характерных признаков и различных специализированных тестов.
Окончательным тестом на АФ в настоящее время является тест на разрыв хромосом: некоторые клетки крови пациента обрабатывают в пробирке химическим веществом, которое сшивает ДНК. Нормальные клетки способны исправить большинство повреждений и не подвергаются серьезному воздействию, тогда как при заболевании клетки демонстрируют заметное разрушение хромосом. Для этого теста обычно используются два химических вещества: DEB (диэпоксибутан) и MMC (митомицин C). Эти тесты могут быть выполнены пренатально на клетках из ворсин хориона или из околоплодных вод.
Анализы крови могут быть выполнены, чтобы определить уровни красных и белых клеток крови и тромбоцитов. Рентгенологическое исследование может выявить наличие и степень пороков развития скелета и внутренних структурных аномалий.
Многие случаи анемии Фанкони вообще не диагностируются или не диагностируются своевременно. АФ следует заподозрить и проверить на наличие у любого ребенка, родившегося с аномалиями большого пальца и руки, описанными ранее. Любой, у кого развивается апластическая анемия в любом возрасте, должен пройти тест на АФ, даже если других дефектов нет. Любой пациент, у которого в раннем возрасте развивается плоскоклеточный рак головы и шеи, желудочно-кишечного тракта или гинекологической системы с или без употребления табака или алкоголя, должен пройти обследование на АФ. Многие пациенты с АФ не показывают никаких других отклонений. Необходимо провести тест на АФ, прежде чем рассматривать трансплантацию стволовых клеток на предмет апластической анемии или лечения рака, поскольку стандартные протоколы химиотерапии и облучения могут оказаться токсичными для пациентов с заболеванием.
Молекулярно-генетическое тестирование доступно для всех 18 генов, связанных с АФ. Тестирование комплементации обычно проводится первым, чтобы определить, какой ген мутирует. Затем можно провести анализ последовательности соответствующего гена, чтобы определить конкретную мутацию в этом гене. Если мутация не идентифицирована, клинически доступен анализ делеции/дупликации генов.
Целевой мутационный анализ доступен для обычной ашкеназской еврейской мутации FANCC.
— Клиническое исследование.
Чтобы определить степень заболевания у индивидуума с диагнозом АФ, при необходимости рекомендуется следующие исследования:
- Ультразвуковое исследование почек и мочевыводящих путей.
- Формальный тест слуха.
- Оценка развития (особенно важно для малышей и детей школьного возраста).
- Обращение к офтальмологу, отоларингологу, эндокринологу, хирургу руки, гинекологу (для женщин, как указано), гастроэнтерологу, урологу, дерматологу, ЛОР-хирургу, генетическому консультанту.
- Оценка гематологом, включая полный анализ крови, гемоглобина плода и аспирата костного мозга для морфологии клеток и исследования хромосом (цитогенетика), а также биопсию для определения клеточности.
- HLA-типирование отдельных лиц, братьев и сестер и родителей для рассмотрения трансплантации гемопоэтических стволовых клеток.
- Полное типирование крови.
- Химия крови (оценка состояния печени, почек, щитовидной железы, липидов и железа).
Лечение анемии Фанкони
Лечение анемии Фанкони направлено на конкретные симптомы, которые проявляются у каждого человека. Лечение может потребовать скоординированных усилий команды специалистов. Педиатрам, хирургам, кардиологам, почечным специалистам (нефрологам), урологам, гастроэнтерологам, специалистам, которые оценивают и лечат проблемы со слухом (аудиологам и отоларингологам), глазным специалистам и другим специалистам здравоохранения, возможно, потребуется систематически и всесторонне планировать лечение пострадавшего.
Рекомендации по лечению были согласованы на консенсусной конференции 2014 года (https://www.nhlbi.nih.gov/health/health-topics/topics/fanconi/).
- Введение андрогенов (мужской гормон): андрогены улучшают показатели крови примерно у 50% людей с АФ. Самый ранний ответ наблюдается в эритроцитах, при этом увеличение гемоглобина обычно происходит в течение первого или двух месяцев лечения. Ответы на количество белых клеток и количество тромбоцитов являются переменными. Реакции тромбоцитов, как правило, неполные и не могут быть замечены до нескольких месяцев терапии. Улучшение, как правило, самое большое для количества эритроцитов. Сопротивление терапии может развиваться со временем.
- Гематопоэтические факторы роста: гранулоцитарный колониестимулирующий фактор (G-CSF) может улучшать количество нейтрофилов у некоторых людей. Обычно используется только для поддержки при интеркуррентных заболеваниях.
- Гематопоэтическая трансплантация стволовых клеток (HSCT): единственная лечебная терапия для гематологических проявлений АФ. Донорские стволовые клетки могут быть получены из костного мозга, периферической крови или пуповинной крови.
- Лечение рака: Лечение злокачественных новообразований является сложной задачей в связи с повышенной токсичностью, связанной с химиотерапией и облучением при анемии Фанкони. Следует проявлять осторожность в центрах, имеющих опыт лечения пациентов с АФ.
Хирургическое вмешательство может быть необходимо для коррекции пороков развития скелета, таких как поражения больших пальцев рук и костей предплечья, пороки сердца и желудочно-кишечные нарушения, такие как трахеопищеводный свищ или атрезия пищевода, а также анальная атрезия.
Некоторые химические вещества могут увеличивать риск хромосомных нарушений у людей с АФ, и их следует избегать, когда это возможно. Эти химические вещества включают табачный дым, формальдегид, гербициды и органические растворители, такие как бензин или разбавитель краски.
Рекомендуется генетическое консультирование для пострадавших людей и их семей.
Источник