Апластическая анемия фанкони лечение

Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Анемия Фанкони была впервые описана в 1927 г. швейцарским педиатром Гвидо Фанкони, который сообщил о 3 братьях с панцитопенией и физическими пороками. Термин «анемия Фанкони» был предложен Негели в 1931 г. для обозначения комбинации семейной анемии Фанкони и врождённых физических пороков. Сегодня для установления диагноза анемии Фанкони не обязательно наличие ни пороков развития, ни анемии Фанкони как таковой. Анемия Фанкони – редкое аутосомно-рецессивное заболевание, её частота составляет 1 на 360 000 родившихся детей с соотношением 1,1:1 в пользу мальчиков.
К настоящему времени известно о более чем 1200 случаях анемии Фанкони и их количество быстро увеличивается в результате внедрения методов лабораторной диагностики, позволяющих установить диагноз заболевания у сиблингов больного анемией Фанкони ещё до манифестации апластической анемии, а также у больных с характерными пороками развития, но без гематологических аномалий.
[1], [2], [3], [4], [5]
Причины анемии Фанкони
Анемия Фанкони – аутосомно-рецессивное заболевание с вариабельной пенетрантностью и генетической гетерогенностью. Гетерозиготное носительство встречается с частотой 1:300. При кариотипировании лимфоцитов и фибробластов больных анемией Фанкони обнаруживают в большом проценте случаев хромосомные аномалии. Считается, что дефектные гены, ответственные за снижение репаративных свойств организма, расположены в 22-й и 20-й хромосомах.
[6], [7], [8], [9]
Патогенез анемии Фанкони
В костном мозге выявляют пониженную клеточность, угнетение всех ростков кроветворения (эритроидного, миелоидного, мегакариоцитарного), разрастание жировой ткани. Дефект гемопоэза при анемии Фанкони локализован на уровне стволовой клетки. Кроветворные клетки имеют повышенное время созревания. Длительность жизни эритроцитов детей с анемией Фанкони существенно снижена (в 2,5-3 раза).
Симптомы анемии Фанкони
Средний возраст установления диагноза анемии Фанкони составляет 7,9 лет среди мальчиков и 9 – среди девочек, причём 75% случаев анемии Фанкони диагностируют в период с 3 до 14 лет. Настороженность в отношении анемии Фанкони ни в коем случае не должна ограничиваться возрастными рамками: вариации возраста пациентов, в котором устанавливался диагноз, необычайно широки – от рождения до 48 лет и от рождения до 32 лет для лиц женского и мужского пола соответственно.
Классический облик больного анемией Фанкони – низкий рост, микроцефалия, микрофтальмия, смуглый оттенок кожи («постоянный загар»), участки гипер- и гипопигментации кожи и слизистых оболочек и уродливые I пальцы рук. При анемии Фанкони различные органы и системы поражаются врождёнными пороками и аномалиями развития в неравной степени. Около 6% больных вообще не имеют никаких аномалий. Ранее такие случаи описывались в литературе под названием анемии Эстрена-Дамешека – по имени авторов, которые в 1947 г. описали 2 семьи с конституциональной гипопластической анемией без пороков развития. Диагноз анемии Фанкони должен быть обязательно подтверждён тестами на гиперчувствительность хромосом, тем более что аномалии развития могут быть общими для анемии Фанкони и других наследственных апластических анемий, например, врождённого дискератоза. Выраженность пороков развития может сильно варьировать даже в пределах одной семьи: известно множество случаев анемии Фанкони среди сиблингов, у одного из которых не было пороков развития, а у другого были.
Лабораторные признаки анемии Фанкони
Трёхростковая аплазия выступает наиболее типичной манифестацией анемии Фанкони, однако наблюдения за инициально гематологически интактными гомозиготами показали, что зачастую тромбоцито- или лейкопения предшествуют развитию панцитопении. Первые гематологические аномалии при анемии Фанкони закономерно обнаруживают после респираторных вирусных инфекций, прививок, иногда гепатитов – так, как это характерно и для идиопатических апластических анемий. Для анемии Фанкони даже в доанемическую фазу типичен выраженный макроцитоз, сопровождающийся значительным повышением уровня фетального гемоглобина. Пунктат костного мозга, как правило, обеднён кроветворными клеточными элементами, преобладают лимфоциты, встречаются плазматические, тучные клетки и стромальные элементы – клиническая картина, неотличимая от идиопатической апластической анемии. Зачастую в аспирате костного мозга обнаруживают дисмиелопоэз и дизэритропоэз, в частности, мегалобластоидность, благодаря которой Фанкони назвал эту анемию «пернициозиформной». В биоптатах костного мозга на ранних стадиях заболевания выявляются гиперклеточные участки активного резидуального гемопоэза, которые исчезают по мере прогрессирования заболевания.
Один из фундаментальных феноменов, характерных для клеток крови больных анемией Фанкони, – это их склонность к формированию специфических хромосомных аномалий – разрывов, сестринских обменов, эндоредупликаций при культивировании клеток in vitro. Инкубация ФГА-стимулированных лимфоцитов больных анемией Фанкони с бифункциональными алкилирующими агентами, которые вызывают сшивки ДНК между гуанидиновыми основаниями, расположенными как на одной, так и на двух комплементарных цепях – нитроген-мустардом, препаратами платины, митомицином и особенно диэпоксибутаном – резко увеличивает количество аберраций. Этот феномен, получивший название кластогенного эффекта, лежит в основе современной диагностики и дифференциальной диагностики анемии Фанкони, поскольку спонтанные аберрации могут как отсутствовать у больных анемией Фанкони, так и присутствовать у больных с другими синдромами, в частности с синдромом Ниймеген. Под влиянием бифункциональных алкилирующих агентов происходит замедление клеточного цикла: клетки больных анемией Фанкони останавливаются в G2 фазе митотического цикла, что послужило основанием для разработки ещё одного диагностического теста для анемии Фанкони с помощью метода проточной флюориметрии.
Возраст первого появления анемии Фанкони в одной семье часто конкордантен, но может и существенно варьировать, в том числе и у однояйцевых близнецов. В прошлом при отсутствии специфического лечения (андрогены или трансплантация костного мозга) и проведении только гемотрансфузий заболевание неуклонно прогрессировало: 80% больных умирали от осложнений панцитопении в течение 2 лет после установления диагноза апластической анемии и практически все больные умирали через 4 года. Необходимо упомянуть, что зафиксировано несколько случаев спонтанного улучшения и даже полного восстановления гематологических показателей.
Вторыми по частоте развития гематологической презентацией анемии Фанкони выступают острые лейкозы и миелодиспластические синдромы. Примерно у 10% больных анемией Фанкони, клинические случаи которых описаны в литературе, впоследствии развился острый лейкоз. Во всех случаях, за исключением 2, лейкозы были миелоидными. Описаны даже случаи установления диагноза анемии Фанкони у пациента с резидуальной цитопенией через много лет после успешной химиотерапии ОМЛ. Несколько ниже частота развития миелодиспластических синдромов – около 5%, причём только у 1/5 из этих больных прослежена дальнейшая эволюция МДС в ОМЛ и несколько больных с МДС прожили более 10 лет. Согласно исследованиям Международного регистра анемии Фанкони риск развития ОМЛ или МДС у больных анемией Фанкони равен 52% к 40 годам. Зачастую выявляют кариотипические аномалии (моносомию 7, трисомию 21, делецию 1), которые позволяют квалифицировать ОМЛ и МДС у больных анемией Фанкони как вторичные. Интересно, что, хотя риск развития МДС/ОМЛ у больных с хромосомными аномалиями примерно в 10 раз выше, чем без таковых, наличие хромосомных аберраций не означает обязательного развития МДС. Клоны, несущие аномалии, могут спонтанно исчезать или сменять друг друга.
Кроме гематологических аномалий для больных анемией Фанкони характерна склонность к развитию опухолей. Риск развития злокачественных опухолей у больных анемией Фанкони составляет 10%, из них 5% приходится на долю опухолей печени и 5% – на остальные опухоли. Опухоли реже встречаются у детей – средний возраст диагностики опухолей печени составляет 16 лет, а остальных опухолей – 23 года. Опухоли печени (гепатоцеллюлярная карцинома, гепатома, аденома и др.), а также пелиоз («кровяные озерца») встречаются чаще у мужчин (соотношение 1,6:1), причём применение андрогенов увеличивает риск их возникновения. В то же время внепечёночные опухоли чаще встречаются у женщин (соотношение 3:1) даже после исключения опухолей гинекологической сферы. Самые частые формы рака при анемии Фанкони – чешуйчатоклеточные карциномы языка и рак пищевода, на которые приходится более 30% всех внепечёночных опухолей при анемии Фанкони, остальные опухоли встречаются в 5-7 раз реже.
Какие анализы необходимы?
Лечение анемии Фанкони
Как уже было указано, симптоматическое лечение апластической анемии при анемии Фанкони не способно коренным образом изменить прогноз заболевания. Первой и единственной на сегодняшний день группой препаратов, позволяющих улучшить кратко- и среднесрочный прогноз при анемии Фанкони, являются андрогены. Впервые они с успехом были применены для лечения анемии Фанкони Shahidi и Diamond в 1959 г. На Западе самым популярным андрогеном с относительно приемлемыми побочными эффектами служит оксиметалон (доза 2-5 мг/кг), в Украине по-прежнему используют метандростенолон (доза 0,2-0,4 мг/кг). При лечении андрогенами гематологический ответ различного качества достигается примерно у 50% больных. Эффект от андрогенов проявляется через 1-2 мес, затем происходит подъём уровня лейкоцитов, а в последнюю очередь увеличивается число тромбоцитов, причём для достижения плато ответа тромбоцитов нередко требуется 6-12 мес. При отмене андрогенов практически у всех пациентов происходит рецидив заболевания, отсутствие рецидива панцитопении после отмены андрогенов описано лишь у небольшого числа больных и, как правило, было связано с наступлением пубертата. Именно поэтому после достижения максимума гематологического улучшения дозу андрогенов следует осторожно снижать, не отменяя её совсем. Применение андрогенов значимо увеличивает продолжительность жизни у ответивших на лечение пациентов: медиана продолжительности жизни составляет 9 лет после установления диагноза против 2,5 лет соответственно для тех пациентов, у которых лечение андрогенами не было эффективным. Ранее в целях предотвращения несвоевременного закрытия зон роста совместно с андрогенами назначали преднизолон в дозе 5-10 мг через день, однако самостоятельного значения в лечении анемии Фанкони глюкокортикостероиды не имеют.
На настоящий момент единственным методом окончательного излечения гематологического синдрома при анемии Фанкони служит аллогенная трансплантация гемопоэтических стволовых клеток (ТГСК). Всего в мире проведено более 250 трансплантаций гемопоэтических клеток по поводу анемии Фанкони.
Особую сложность представляет проблема лечения лейкемий и миелодиспластических синдромов у больных анемией Фанкони, поскольку повышенная чувствительность тканей этих больных ко многим химиотерапевтическим агентам и сниженный костномозговой резерв предрасполагают к развитию тяжёлой висцеральной и гематологической токсичности. К настоящему времени подавляющее большинство из более 100 больных анемией Фанкони с лейкемиями и миелодиспластическими синдромами погибло. Как правило, смерть наступает в течение 2 мес после установления диагноза лейкемии, хотя случаи диагностики анемии Фанкони через много лет после успешного лечения острого лейкоза говорят по крайней мере о теоретической возможности успешной химиотерапии. Более оптимистичен прогноз у больных ОМЛ и МДС, которым была проведена аллогенная ТГСК без предшествующей химиотерапии.
Какой прогноз имеет анемия Фанкони?
Без успешной трасплантации костного мозга анемия Фанкони имеет серьезный прогноз. При этом больные больше страдают и чаще погибают не от анемии, а от оппортунистических инфекций из-за нейтропении и дефекта иммунитета или повышенной кровоточивости из-за тромбоцитопении. Дети с анемией Фанкони имеют повышенный риск развития нелимфоидной лейкемии (5-10%).

Анемия Фанкони (АФ), или панцитопения Фанкони, является синдромом нестабильности генома. Это редкое наследственное аутосомно-рецессивное заболевание с вариабельной пенетрантностью и генетической гетерогенностью. АФ была впервые описана в 1927 г. швейцарским педиатром Гвидо Фанкони, который сообщил о 3 братьях с панцитопенией и пороками физического развития. Термин «анемия Фанкони» был предложен Негели в 1931 г. для обозначения комбинации семейной анемии Фанкони и врожденных физических пороков. По настоящее время описано чуть более 2000 случаев АФ. Частота гетерозиготного носительства существенно различается в разных популяциях. Традиционно указывалась цифра 1:300, по последним данным североамериканского регистра, она составляет 1:181, в Израиле — 1:93. Следует заметить, что около 6 % больных не имеют никаких аномалий развития. Некоторые мутации этнически закреплены, в основе их распространения лежит «эффект основателя» — потеря генетической вариабельности в популяциях, основанных малым количеством предков, что свойственно для относительно небольших популяций. Эти мутации встречаются у евреев-ашкенази, испанских цыган, голландцев, выходцев с Канарских островов, у жителей Южной Африки и Кореи. В некоторых из этих популяций частота носительства этнически закрепленных мутаций довольно высока и оценена приблизительно как 1:100. Частота встречаемости тех или иных мутаций в РФ не изучена.
Этиология и патогенез
Нарушение структуры ДНК является результатом воздействия как внутренних (депуринизация, дезаминирование, воздействие эндогенных альдегидов, особенно формальдегида, и активных форм кислорода), так и внешних факторов (ионизирующее и УФ облучение, химические мутагены). При воздействии этих факторов происходят реакции алкилирования, окисления, восстановления, связывания с формальдегидными группами азотистых оснований. В итоге возникают изменения одного или нескольких оснований, вставки и делеции, образование тиминовых димеров, одно- и двухцепочечные разрывы ДНК, образование сшивок между основаниями одной цепи или комплементарными цепями ДНК, между ДНК и белковыми молекулами. Несмотря на это, в целом геном остается свободным от «ошибок», так как клетка имеет механизмы детекции и репарации поврежденной ДНК. Поврежденное основание может быть восстановлено непосредственно его заменой или обратной химической реакцией (direct repair), в других случаях необходимы более сложные процессы, обеспечивающие удаление поврежденного участка ДНК и достраивание правильной последовательности с использованием комплементарной цепи, редко — гомологичной хромосомы. Такая репарация ДНК — сложный многоступенчатый процесс взаимодействия нескольких каскадных путей. Процесс репарации происходит на разных этапах клеточного цикла. При АФ нарушается способность клетки исправлять определенный тип повреждений ДНК — поперечные межхроматидные сшивки (DNA interstrand crosslink), которые препятствуют работе репликационной вилки. Поперечные межхроматидные сшивки формируются как под воздействием продуктов естественного метаболизма клетки (в первую очередь эндогенных альдегидов, но также и активных форм кислорода), так и под воздействием химических веществ, в частности химиотерапевтических препаратов. К такому типу химических веществ относятся алкилирующие соединения, имеющие в своем составе две активные алкильные группы, обеспечивающие им активное связывание с определенными основаниями: цисплатин, митомицин С, азотистый иприт, псорален, диэпоксибутан, мелфалан, циклофосфамид, мустарген, стрептозоцин. Протеины, функция которых нарушается при АФ, задействованы во всех этапах репарации межхроматидной поперечной сшивки. Этот сложный многоступенчатый процесс получил название FA-pathway, а протеины, задействованные в нем, — АФ-протеины. Ключевую роль в этом процессе играет моноубиквитинирование гена FANCD2, который координирует процессы вырезки поврежденных нуклеотидов, прямое достраивание поврежденного участка и гомологичную рекомбинацию. При АФ клетка не способна адекватно исправлять повреждения ДНК, накопление поломок которой может приводить к костномозговой недостаточности, аномалиям физического развития и предрасположенности к развитию опухолей.
Спектр мутаций, которые приводят к АФ:
1) FANCA.
Мутации в этом гене — самые распространенные и встречаются в 60–70 % случаев АФ. Известно более 100 мутаций, из которых около трети приходится на точечные, еще треть представляют собой микроделеции, и около 40 % представлены крупными делециями. Описаны также и малые дупликации. По действию мутации в гене FANCA могут быть гипоморфными, т. е. приводить к частичной потере функции белка; они характеризуются более мягкими клиническими проявлениями, однако большая часть мутаций вызывает полную потерю функции. Ряд мутаций в гене FANCA имеет повышенную частоту распространения. Так, микроделеция в 38-м экзоне с.3788_3790delTCT — самая распространенная мутация при АФ в мире (20,7 % всех аллелей с мутацией). При этом она встречается у 80 % пациентов с АФ с Канарских островов, где частота встречаемости АФ достигает 1:16000 новорожденных. Кроме того, эта мутация встречается в 51 % случаев АФ в Бразилии. Для подтверждения «эффекта основателя» для данной мутации был проведен анализ гаплотипа пациентов путем изучения вариабельных тандемных повторов и однонуклеотидных полиморфизмов гена FANCA у 28 пациентов с мутацией с.3788_3790delTCT из различных частей света. Все, за исключением одного пациента, имели общего предка. По всей видимости, Канарские острова послужили местом происхождения и распространения заболевания из Европы в Америку, так как несколько веков назад практически все суда из Испании в Америку шли через Канарские острова. Однако, учитывая, что эта мутация тем не менее составляет 2–5 % всех FANCA-мутаций, существует также и мнение, что она связана с явлением существования определенных участков генома с повышенной мутационной способностью, так называемых hotspot. Другой пример «эффекта основателя» — мутация с.295С>Т, которую выявляют почти во всех случаях АФ у испанских цыган. При этом носительство этой мутации среди испанских цыган определено как 1:67. Функционально и фенотипически варианты мутаций в гене FANCA проявляются одинаково.
2) FANCC.
Мутации в гене FANCC встречаются в 10–15 % случаев АФ, почти 90 % случаев представлено двумя мутациями — с.711+4А>Т и delG332. Самая частая мутация в гене FANCC — с. 711+4А>Т, ее выявляют в гомозиготном состоянии в 80 % случаев АФ у евреев-ашкенази. Частота гетерозиготного носительства этой мутации среди евреев-ашкенази достигает 40 %. При этом встречаются и спорадические случаи. Генотип FANCC с.711+4А>Т также распространен среди больных АФ в Японии. В Нидерландах более чем 50 % случаев АФ — это гомозиготные носители мутации с.67delG (также известна как delG332), приводящей к сдвигу рамки считывания в гене FANCC.
3) FANCG.
FANCG задействован в 10 % случаев АФ. Встречаются практически все типы мутаций, за исключением крупных делеций. Клинически характеризуются более частым и быстрым развитием миелодисплазии или лейкоза. Встречаются как спорадические случаи, так и этнически ассоциированный вариант — около 80 % случаев АФ у чернокожих южноафриканцев (Bantu-speakers) (Южная Африка, Свазиленд, Малави и Мозамбик) имеют мутацию 637_643delTACCGCC. Заболевание характеризуется частыми нарушениями пигментации кожи, слабовыраженными аномалиями развития и сравнительно поздними, но тяжелыми гематологическими проявлениями, обусловленными в том числе и поздним обращением к врачу.
Развитие костномозговой недостаточности связывают с повышенным апоптозом гемопоэтических клеток, однако истинные патогенетические механизмы костномозговой недостаточности при АФ мало изучены из-за сложности получения адекватной биологической модели развития заболевания. Последние исследования на ксенографтных моделях и in vitro показали, что в ответ на накопление нерепарированных повреждений ДНК происходит активация р53 проапоптотического пути и запуск поздней р21(Cdkn1a)-зависимой блокировки клеточного цикла в фазе G0/G1 с последующей элиминацией ранних гемопоэтических предшественников из костного мозга. Этот механизм запускается в пренатальном периоде на этапе формирования пула стволовых клеток и ранних клеток-предшественников гемопоэза, что приводит к значительному снижению их количества. Накопление дефектов ДНК после рождения в результате различных физико-химических воздействий усугубляет нарушение гемопоэза. Кроме выраженного апоптоза ранних клеток-предшественников происходит нарушение базовых свойств стволовых кроветворных клеток — способности к самоподдержанию собственной популяции, пролиферации и дифференцировке в различные линии гемопоэза. Генетическая нестабильность при АФ реализуется в повышенной частоте развития ряда опухолей, наиболее частые — ОМЛ и плоскоклеточный рак кожи головы и шеи, слизистых оболочек рта и мочеполового тракта — то есть тканей, характеризующихся высокой пролиферативной активностью.
Клиническая картина
Признаки и симптомы, а также частота их встречаемости указаны в таблице 1.
Таблица 1 | Признаки и симптомы анемии Фанкони
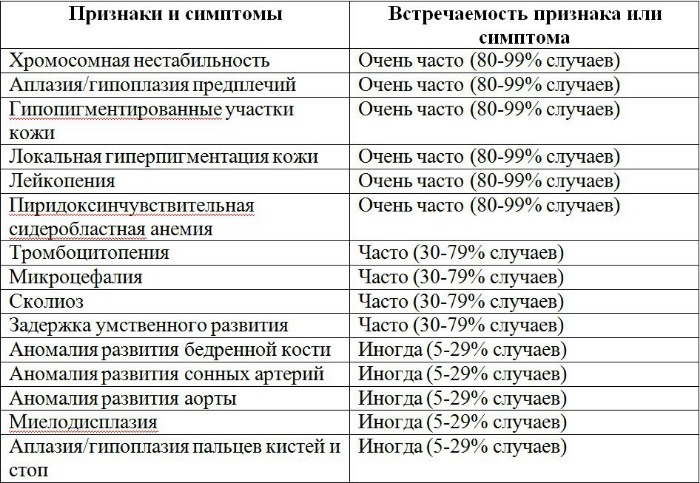
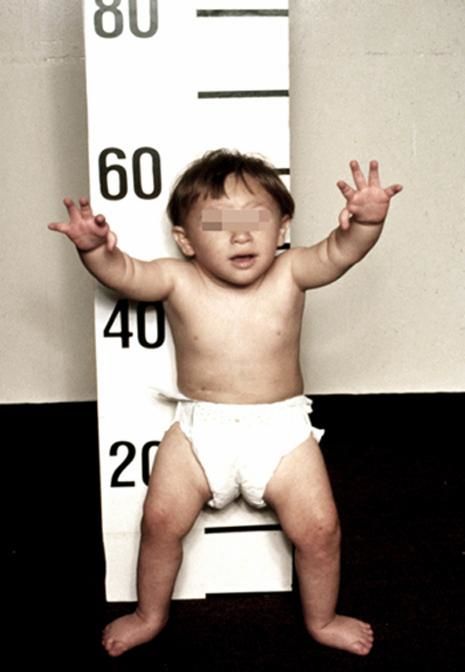
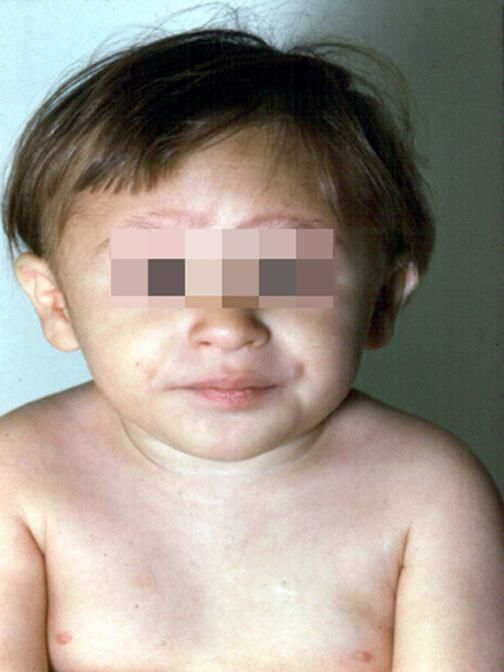
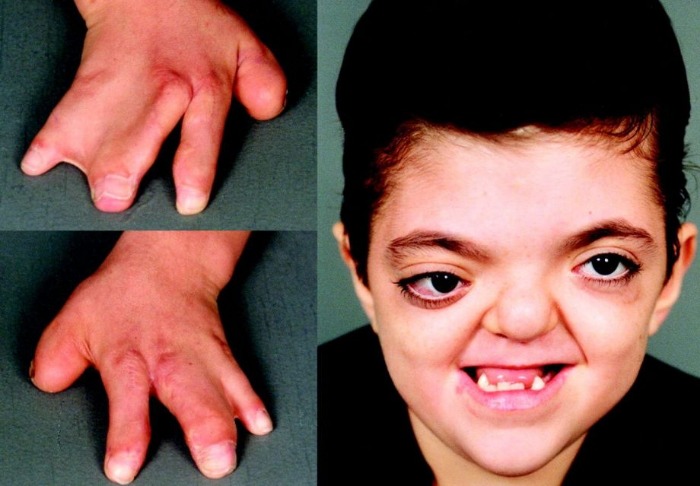
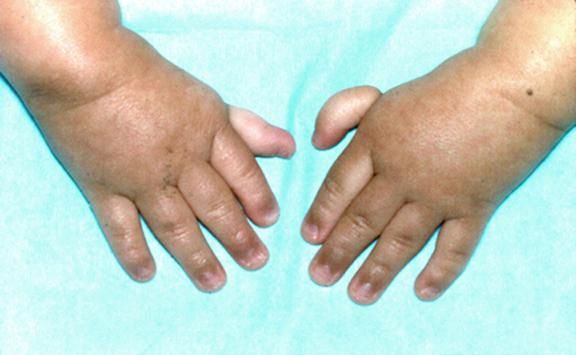
Рисунки 1–5 | Пороки развития, наблюдаемые при анемии Фанкони.
Диагностика
Помимо тщательного физикального осмотра, сбора семейного анамнеза, общего анализа крови и пункции костного мозга для более точной диагностики используются такие методы лабораторной диагностики, как тест на ломкость хромосом, метод MLPA, высокопроизводительное секвенирование и секвенирование по Сэнгеру.
1) Тест на ломкость хромосом.
«Золотым стандартом» скрининга для выявления АФ был и остается тест с диэпоксибутаном (1,3-butadienediepoxide) и его вариант с митомицином С (MMC). Еще в начале изучения АФ было отмечено, что фибробласты и лимфоциты больных АФ в культуре клеток демонстрируют спонтанную повышенную ломкость хромосом. Значительные различия в уровне спонтанных аберраций у больных (вплоть до отсутствия таковых) требовали унифицированного и точного метода детекции. Позже была показана повышенная чувствительность клеток больных АФ к действию алкилирующих агентов, вызывающих поперечные сшивки между нуклеотидами, что препятствует образованию нормальной репликативной вилки для запуска процесса репарации ДНК, позже получивших общее название interstrand cross-link agent. На основании этого был предложен цитогенетический метод диагностики АФ: после обработки лимфоцитов или фибробластов алкилирующим веществом (в нелетальной для клеток концентрации) определяют частоту и спектр спонтанных и индуцированных in vitro хромосомных аберраций. Обычно ставится несколько параллельных клеточных культур, стимулированных фитогемагглютинином лимфоцитов периферической крови: без добавления алкилирующего агента (для определения спонтанного уровня аберраций) и с его добавлением в разной концентрации. Затем в метафазных пластинках подсчитывают число хромосомных разрывов. Для АФ характерны разрывы хромосом с образованием радиальных фигур, фрагментов, хромосомных и хроматидных разрывов, а также три- и тетрарадикалов. При анализе результатов учитывают число разрывов по отношению к числу проанализированных метафаз, процент клеток с разрывами и ряд других показателей. Необходимо отметить, что значения, при которых тест на ломкость хромосом считается положительным, в различных лабораториях варьируются. Тест не имеет 100 % специфичности. Положительный тест на ломкость хромосом бывает у пациентов с синдромом Ниймеген (мутации в гене NBS1), синдромом Робертса (мутации в гене ESCO2), Warsaw breakage syndrome (мутации в гене DDX11), синдромом Блюма (ген BLM), врожденном дискератозе и некоторых других синдромах. В то же время АФ-подобные синдромы могут оказаться действительно АФ с соответствующим генетическим дефектом.
2) Секвенирование по Сэнгеру долгие годы было основным методом определения мутаций при АФ. Учитывая значительные размеры генов, секвенирование каждого из них представляет собой довольно трудоемкий и дорогостоящий процесс. Его обычно проводили после анализа на группу комплементации, предварительно определив вероятный ген. Анализ нуклеотидной последовательности для всех известных АФ-генов затруднен количеством возможных мутаций в каждом, их разнообразием, в том числе в виде крупных инсерций или делеций (indel-мутации). Их длина может варьировать от одного до нескольких сотен и даже тысяч нуклеотидных оснований, что подразумевает использование совершенно разных методов молекулярно-генетического исследования.
3) Метод MLPA (мультиплексная амплификация лигазносвязанных проб) предназначен для определения делеций и амплификаций определенных последовательностей гена длиной до нескольких десятков нуклеотидов. Одновременно может быть исследовано до 60 таких последовательностей, что позволяет выявить как сравнительно небольшие делеции, так и делеции отдельных экзонов и целого гена. Метод MLPA используют для инициального скрининга делеции в гене FANCA, параллельно ген FANCA полностью секвенируют. Если пациент мужского пола, его исследуют на наличие делеций ген FANCB. Выявленные делеции желательно подтвердить другим методом. При этом методы, которые могут быть использованы, требуют индивидуальной разработки в каждом конкретном случае: количественная ПЦР, ПЦР длинных фрагментов (long range PCR) и хромосомный микроматричный анализ.
4) Высокопроизводительное секвенирование.
Метод, который позволяет одномоментно анализировать от нескольких генов до полного генома, является наиболее подходящим для определения мутаций при АФ. Возможны несколько вариантов теста. Первый — секвенирование экзома, позволяет получить максимальный объем информации. Второй — секвенирование ограниченного числа интересующих и уже описанных в литературе генов (таргетное ресеквенирование), список которых можно дополнять или моделировать в соответствии с потребностями исследования. Современные коммерческие панели генов, как правило, помимо генов АФ включают большое число генов, ответственных за развитие других врожденных синдромов, в том числе и АФ-подобных. Внедрение в практику высокопроизводительного секвенирования позволяет избежать последовательного трудоемкого исследования каждого из известных генов методом секвенирования по Сэнгеру, однако пока не позволяет с должной уверенностью выявить крупные делеции и дупликации. Найденные мутации требуют подтверждения одним из других подходящих методов. При обнаружении новых мутаций необходимо подтверждение их патогенности в функциональном тесте и др.
Пренатальная и преимплантационная диагностика должна проводиться в первую очередь в семьях, где ранее были установлены патогенетические мутации. В этом случае проводится целенаправленный поиск известной мутации. Материалом для диагностики служат клетки плода, получаемые путем биопсии ворсин хориона на 10–12 неделе беременности. Следует помнить, что генетический анализ занимает не менее 2–3 недель. Если нет возможности провести молекулярно-генетическое исследование, возможно выполнение теста на ломкость хромосом клеток ворсин хориона на 10–12 неделе беременности либо при амниоцентезе на 15–18 неделе. Однако молекулярно-генетическое исследование предпочтительнее.
Лечение
Основной метод лечения АФ — аллогенная трансплантация гемопоэтических стволовых клеток.
Из медикаментозного лечения в настоящий момент применяют андрогены. Препараты этой группы (оксиметалон, метандростенолон) позволяют достичь гематологического ответа примерно у 50 % больных. Также применение андрогенов значимо увеличивает продолжительность жизни у ответивших на лечение пациентов: медиана продолжительности жизни составляет 9 лет после установления диагноза против 2,5 лет соответственно для тех пациентов, у которых лечение андрогенами не было эффективным.
При возникновении сопутствующих заболеваний, например, инфекций, обосновано применение гранулоцитарного колониестимулирующего фактора, который способен временно увеличить количество нейтрофилов и облегчить протекание заболевания.
В 2015 году коллективу ученых удалось успешно применить технологию Crispr/Cas9 для редактирования мутации с.67delG гена FANCC.
Источники:
- https://www.bloodjournal.org/content/126/23/3622?sso-checked=true
- https://www.ncbi.nlm.nih.gov/books/NBK1401/
- https://rarediseases.info.nih.gov/diseases/6425/fanconi-anemia
- https://rarediseases.org/rare-diseases/fanconi-anemia/
- https://cyberleninka.ru/article/v/geneticheskaya-diagnostika-anemii-fankoni-obzor-literatury
Нашли опечатку? Выделите фрагмент и нажмите Ctrl+Enter.