Дэб тест при анемии


Анемия Фанкони – это генетическое заболевание, которое передается по аутосомно-рецессивному типу и характеризуется нарушением кроветворения, формированием злокачественных новообразований, пороками развития, ломкостью хромосом. Проявляется частыми кровотечениями, кровоподтеками на коже, вялостью, бледностью, склонностью к инфекциям. Диагностика проводится лабораторными методами, назначаются цитогенетическое, молекулярно-генетическое и клиническое исследования крови, миелограмма. Основные способы лечения – пересадка костного мозга, медикаментозное поддержание кроветворения, переливание крови.
Общие сведения
Синонимичные названия анемии Фанкони – врожденная панмиелопатия Фанкони, наследственная панмиелопатия. Заболевание названо по фамилии швейцарского педиатра Гвидо Фанкони, который в 1927 году описал врожденную апластическую патологию на основе симптомов у трех братьев. Анемия Фанкони является редкой генетической болезнью, наследуется согласно аутосомно-рецессивному принципу. Эпидемиологические показатели низкие – 1 больной ребенок на 350 тысяч новорожденных. Распространенность одинакова среди представителей женского и мужского пола, выше в сообществах с разрешенными близкородственными браками, например, у некоторых южноафриканских народов.

Анемия Фанкони
Причины
Заболевание является наследственным, развивается при передаче дефектного гена от родителей к ребенку. Выявлено 15 генов, мутации которых проявляются анемией Фанкони. Из них 14 расположены в аутосомах и являются рецессивными, 1 тип гена находится в X-хромосоме (сцепленной с полом). Все эти гены отвечают за производство определенного фермента, участвующего в репарации ДНК.
Аутосомно-рецессивное наследование подразумевает, что и отец, и мать должны быть носителями патологической генетической информации. При этом сами они, как правило, здоровы. Вероятность рождения больного ребенка в такой паре составляет 25%. Генетическая панмиелопатия диагностируется у детей и взрослых, получивших от каждого из родителей один и тот же измененный ген. В крайне редких случаях анемия провоцируется передачей дефектной Х-сцепленной хромосомы. Женщины могут быть носительницами мутации, заболевание проявляется только у мальчиков. Риск развития патологии у сына при наличии у матери мутированного гена – 50%.
Патогенез
В норме в клетках организма существуют специальные ферментные системы, которые исправляют разрывы молекул ДНК, поврежденных в процессе биосинтеза или воздействия химических, физических реагентов. При анемии Фанкони обнаруживается генетический дефект в кластере белков, ответственных за репарацию ДНК, что приводит к повышенной ломкости хромосом. В итоге у пациентов развиваются нарушения функций костного мозга – неоплазии и апластическая анемия. Онкологические заболевания чаще всего представлены острым миелоидным лейкозом – злокачественной опухолью миелоидного ростка крови, провоцирующей накопление измененных белых клеток, подавляющих рост эритроцитов, тромбоцитов и нормальных лейкоцитов. При апластической анемии в результате дисплазии костного мозга резко угнетается рост и созревание всех трех видов клеток крови.
Симптомы анемии Фанкони
Более чем у половины пациентов наблюдаются врожденные аномалии развития внутренних органов и скелета. Костные деформации проявляются специфическим внешним видом: больные низкорослые, с уменьшенным размером головы, отсутствием или заметным укорочением большого пальца на руках, недоразвитием лучевой кости, врожденным вывихом бедра и/или наличием шейного ребра, косолапостью, недоразвитым подбородком («птичьим лицом»). Характерна гиперпигментация кожи в виде светлых и коричневатых пятен.
Неврологические расстройства представлены косоглазием, недоразвитием одного или двух глаз, опущением верхнего века, глазным дрожанием, глухотой, умственной отсталостью. Больные зачастую имеют незрелые половые органы, у них отсутствует одно или оба яичка. К распространенным аномалиям строения органов относятся пороки мочевыделительной системы: удвоение мочеточников или лоханки, подковообразные почки, почечные кисты, смещенное наружное отверстие уретры (гипоспадия). Врожденные пороки сердца включают атрезию трехстворчатого клапана, дефект межпредсердной перегородки, митральный стеноз, дефект межжелудочковой перегородки. Пациенты страдают от почечной и сердечной недостаточности.
Ключевые симптомы связаны с постепенным нарастанием нарушений в работе костного мозга. Чаще они дебютируют в детском возрасте (в 5-10 лет). Из-за снижения количества тромбоцитов развивается повышенная кровоточивость: при ранениях кровь долго не сворачивается, легко возникают носовые кровотечения, выделения при менструациях обильны, на теле обнаруживается много «беспричинных» кровоподтеков. Уменьшение числа эритроцитов проявляется анемией с характерной слабостью, быстрой утомляемостью, головокружениями, обмороками, бледностью кожи, учащенным сердцебиением и одышкой. Недостаток лейкоцитов способствует ухудшению сопротивляемости инфекциям. Впоследствии формируется лейкоз, миелодиспластический синдром, онкологические болезни.
Осложнения
Наиболее распространенным осложнением считаются частые инфекционные заболевания. У пациентов развивается ОРВИ, ангина, ринит, бронхит, грипп, тиф, герпес. Рецидивирующий характер болезней и их тяжелое течение приводят к деструкции органов, сопровождаются риском сепсиса. Другим осложнением наследственной анемии являются злокачественные новообразования – лейкемия, эпителиальные опухоли органов шеи и головы, половых органов. Рак у таких больных тяжело поддается лечению из-за повышенной ломкости и сниженной репарации ДНК. Это явление ограничивает применение лучевой терапии, цитотоксических препаратов. Нарушение свертываемости становится причиной больших кровопотерь.
Диагностика
Обследование больных проводят онкологи, гематологи, педиатры, врачи-генетики. Диагностика начинается с анализа анамнестических данных и жалоб. Врач выясняет, имеется ли данное наследственное заболевание у близких родственников, уточняет время появления первых признаков болезни, ранние обращения к врачам. При осмотре оценивает общее состояние пациента, выявляет наличие аномалий развития, гиперпигментированных пятен, кровотечений, кровоподтеков. В большинстве случаев не составляет труда обнаружить типичные деформации костей, недоразвитие глаз. Для подтверждения диагноза и различения анемии Фанкони с приобретенной анапластической анемией проводится ряд лабораторных исследований:
- Клинический анализ крови. Характерны изменения клеточного состава крови. На ранних этапах нарушения кроветворения диагностируется тромбоцитопения и лейкопения, на более поздних – панцитопения (резкое снижение объема эритроцитов, лейкоцитов и тромбоцитов). Возможен умеренный гемолиз без гипербилирубинемии, но с ретикулоцитозом. Значение СОЭ увеличено до 60-80 мм/ч.
- Цитогенетическое исследование клеток. Выполняется проба с диэпоксибутаном, митомицином C, указывающая на частоту и спектр хромосомных аберраций. В пользу генетической анемии рассматриваются показатели ДЭБ-теста более 45%, пограничный уровень – 11-45% (процент клеток с хромосомными разрывами).
- Молекулярно-генетический анализ клеток. Исследуются гены, мутации в которых могут привести к развитию заболевания. В 60-70% случаев мутации обнаруживаются в паре генов FANCA, в 14% – в аллели FANCC, в 10% – в генах FANCG. Частота мутаций в других парах – 0,2-3%.
- Миелограмма. По данным исследования определяется увеличение количества плазматических клеток и макрофагов, фагоцитирующих жиры. Содержание недифференцированных клеток – в пределах нормы. Снижена концентрация клеток миелоцитарного ростка, увеличен показатель лимфоцитов.
Лечение анемии Фанкони
Основная терапия направлена на восстановление процесса кроветворения. Методы лечения подбираются индивидуально, зависят от тяжести заболевания, возраста пациента, наличия и выраженности врожденных аномалий. Дополнительно проводится лечение инфекций и онкопатологий, осуществляются реабилитационные мероприятия. Для устранения анемии используются следующие методы:
- ТКМ. Трансплантация костного мозга является наиболее эффективной в долгосрочной перспективе, но имеет противопоказания, нередко сопровождается развитием осложнений. Оптимальный возраст для проведения операции – до десяти лет. Донорами могут выступать здоровые сестра и братья, подходящие по критериям совместимости. Предварительная интенсивная терапия (кондиционирование) связана с риском токсического воздействия на органы. После трансплантации сохраняется высокая вероятность острого или хронического иммунного конфликта между клетками донора и реципиента.
- Медикаментозная стимуляция кроветворения. При невозможности проведения трансплантации пациентам показано консервативное лечение, временно улучшающее их состояние. Выработка кровяных клеток стимулируется андрогенами (мужскими половыми гормонами) и гематопоэтическими факторами роста – эритропоэтином, фактором стволовых клеток, интерлейкинами-1-12. Параллельно применяются иммунодепрессанты. Медикаментозная терапия способна на протяжении многих лет поддерживать высокое качество жизни больных, но ее эффективность постепенно снижается.
- Переливание компонентов крови. При выраженных побочных эффектах или противопоказаниях к этиотропной терапии (трансплантации, стимуляции кроветворения) назначаются процедуры гемотрансфузии. Переливаются отмытые эритроциты – донорские красные кровяные тельца, освобожденные от поверхностных белков. При кровотечениях и снижении уровня тромбоцитов пациентам вводится тромбоцитарная масса.
Прогноз и профилактика
Продолжительность жизни больных определяется степенью нарушения функции костного мозга. Иногда пациенты доживают до 40 лет без лечения, но нередко умирают в детстве от тяжелой анемии или онкологических заболеваний. Прогноз наиболее благоприятен при своевременном проведении аллогенной трансплантации костного мозга, после которой есть шанс полного восстановления нормального кроветворения и увеличения срока жизни. Поскольку заболевание генетическое, предотвратить его развитие невозможно. Профилактика сводится к медико-генетической консультации супружеских пар из групп риска, планирующих беременность, а также к проведению пренатальной диагностики патологии, в ходе которой из пуповинной вены плода производится забор крови и выполняется ДЭБ-тест. При его положительном результате рассматривается вопрос о прерывании беременности.
Источник
Анемия Фанкони (АФ) — редкое наследственное аутосомно рецессивное заболевание, характеризующееся недостатком кроветворе ния, аномалиями развития и склонностью к развитию опухолей вследствие нестабильно сти генома.
Частота встречаемости 1:350 000 новорож денных, частота носительства по разным ис точникам 1:181—300 [1, 2].
В настоящее время известно 19 генов, связан ных с развитием АФ, каждый из которых от вечает за синтез определенного протеина, так или иначе участвующего в процессе репара ции ДНК [3, 4, 5].
КЛИНИЧЕСКИЙ ПРИМЕР
В январе 2017 г. в гематологическое отделение МДГКБ поступил мальчик М., 1 год 3 мес.
Анамнез. Ребенок от 1 беременности, род ственный брак (родители двоюродные сибсы). Роды 1 на 41 нед, самостоятельные, воды про зрачные.
Масса 2800 г, рост 49 см.
По ЭхоКГ открытый артериальный проток. Выписан на 5 сут в удовлетворительном состо янии, ребенок находился на грудном вскарм ливании по требованию.
В возрасте 2 мес жизни (с 02.12 по 18.12.2015) находился на лечении в ДГКБ № 9 с диагнозом «галактоземия? Холестатический синдром. Вторичный геморрагический синдром. Суб арахноидальное кровоизлияние. Постгемор рагическая анемия тяжелой степени. Дефи цит веса». В отделении у ребенка отмечалась двухростковая цитопения (эритропения, анемия, тромбоцитопения), гипокоагуляция, гипербилирубинемия. Проводилась терапия свежезамороженной плазмой № 3, двукратно введение эритроцитарной массы, гемостати ческая терапия викасолом и дициноном. Ге моррагический синдром купирован.
Настоящая госпитализация в гематологиче ское отделение МДГКБ в связи с появлением фебрильной лихорадки и продуктивного кашля на фоне агранулоцитоза и тромбоцитопении. При поступлении обращали на себя внимание задержка физического и интеллектуального развития, множественные стигмы дисэмбрио генеза, в том числе гипоплазия дистальной фаланги большого пальца левой руки; блед ность кожных покровов, умеренный гемор рагический синдром в виде петехиальных кровоизлияний и экхимозов на коже лица, конечностей.
В общем анализе крови трехростковая ци топения: эритроциты 2,16×1012/л, Нв 74 г/л, MCV 101 фл, MCH 34 пг, тромбоциты 22×109/л, лейкоциты 2,3×109/л, абсолютное число нейтрофилов 0,299×109/л.
В биохимическом анализе крови: умерен ное увеличение ЩФ (421 при Н до 345 Ед/л), ЛДГ (545 при Н до 430 Ед/л), АСТ (105 при Н до 50 Ед/л), АЛТ (74 при Н до 45 Ед/л), ферритина (433 при Н до 60 мкг/л).
При исследовании клеточного иммунитета небольшое увеличение иммунорегулятор ного индекса.
Коагулограмма — нормокоагуляция. ВИЧ, HCV HBsAg отрицательно.
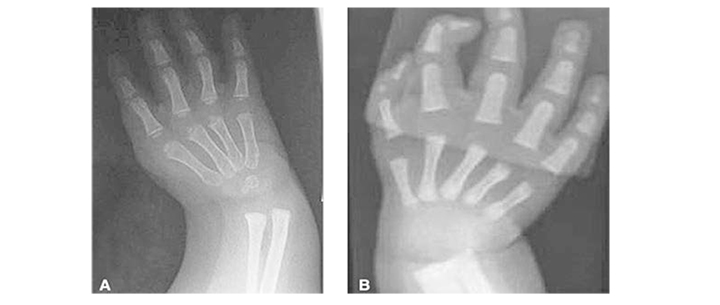
В миелограмме выраженное снижение кле ток эритроцитарного ряда (0,8% при Н 14,5—26,5), сужение нейтрофильного ростка с «обрывом» на уровне миелоцитов, отсутствие мегакариоцитов.
ДЭБтест: положительный (исследовано 30 метафаз, выявлено 144 аберрации; карио тип 46 XY).
ТЕСТ НА ЛОМКОСТЬ ХРОМОСОМ
«Золотым стандартом» скрининга для выявле ния АФ был и остается тест с диэпоксибутаном (ДЭБ) и его вариант с митомицином С (MMC) [5, 6, 7].
Суть метода: после обработки лимфоцитов или фибробластов алкилирующим веществом (в нелетальной для клеток концентрации) определяют частоту и спектр спонтанных и индуцированных in vitro хромосомных абер раций.
Тест не имеет 100% специфичности, лишь очерчивая дифференциальный круг, в кото рый входит АФ, синдром Ниймегена, синдром Робертса, Warsaw breakage syndrome, синдром Блюма, врожденный дискератоз и некоторые другие синдромы. Поэтому в соответствии с рекомендациями по диагностике АФ страте гия генетического скрининга включает так же целевой скрининг мутаций для некоторых популяционных групп, выявление наиболее частых делеций гена FANCA методом MLPA, исследование известных генов АФ методом высокопроизводительного секвенирования. Таким образом, на основании анамнеза, клиниколабораторных данных ребенку был выставлен диагноз «анемия Фанкони».
Ребенку проводилась комплексная терапия, включающая заместительную терапию эри троцитарной взвесью и тромбоконцентра том, антибактериальная, противогрибковая и симптоматическая терапия, на фоне которой анемия, геморрагический синдром, агрануло цитоз, симптомы интоксикации, лихорадки были купированы. Ребенок выписан для даль нейшего амбулаторного наблюдения.
В настоящее время единственным радикаль ным способом лечения АФ является трансплан тация костного мозга от HLAсовместимого донора.
- Glanz A., Fraser F. C. Spectrum of anomalies in Fanconi anemia. J. Med. Genet. 19: 412416, 1982.
- Grompe M., D’Andrea A. Fanconi anemia and DNA repair. Hum. Molec. Genet. 10: 22532259, 2001.
- Auerbach A.D., Rogatko A., SchroederKurth T.M. International Fanconi Anemia Registry: relation of clinical symptoms to diepoxybutane sensitivity. Blood 73: 391396, 1989.
- Brunetti P., Neuci G.G., Vaccaro R., Puxeddu A., Migliorini E. Fanconi’s anaemia. (Letter). Lancet 288: 11941195, 1966. Note: Originally Volume II.
- Joenje H., Patel K.J. The emerging genetic and molecular basis of Fanconi anaemia. Nature Rev. Genet. 2: 446457, 2001.
- Alter B.P. Fanconi’s anaemia and its variability. Brit. J. Haemat. 85: 914, 1993.
- Генетическая диагностика анемии Фанкони. Обзор литературы. Панферова А.В., Тимофеева Н.М., Ольшан ская Ю.В. Онкогематология 3.2016 том 11. DOI 10.17650/1818834620161137685
Источник

Анемия Фанкони (АФ), или панцитопения Фанкони, является синдромом нестабильности генома. Это редкое наследственное аутосомно-рецессивное заболевание с вариабельной пенетрантностью и генетической гетерогенностью. АФ была впервые описана в 1927 г. швейцарским педиатром Гвидо Фанкони, который сообщил о 3 братьях с панцитопенией и пороками физического развития. Термин «анемия Фанкони» был предложен Негели в 1931 г. для обозначения комбинации семейной анемии Фанкони и врожденных физических пороков. По настоящее время описано чуть более 2000 случаев АФ. Частота гетерозиготного носительства существенно различается в разных популяциях. Традиционно указывалась цифра 1:300, по последним данным североамериканского регистра, она составляет 1:181, в Израиле — 1:93. Следует заметить, что около 6 % больных не имеют никаких аномалий развития. Некоторые мутации этнически закреплены, в основе их распространения лежит «эффект основателя» — потеря генетической вариабельности в популяциях, основанных малым количеством предков, что свойственно для относительно небольших популяций. Эти мутации встречаются у евреев-ашкенази, испанских цыган, голландцев, выходцев с Канарских островов, у жителей Южной Африки и Кореи. В некоторых из этих популяций частота носительства этнически закрепленных мутаций довольно высока и оценена приблизительно как 1:100. Частота встречаемости тех или иных мутаций в РФ не изучена.
Этиология и патогенез
Нарушение структуры ДНК является результатом воздействия как внутренних (депуринизация, дезаминирование, воздействие эндогенных альдегидов, особенно формальдегида, и активных форм кислорода), так и внешних факторов (ионизирующее и УФ облучение, химические мутагены). При воздействии этих факторов происходят реакции алкилирования, окисления, восстановления, связывания с формальдегидными группами азотистых оснований. В итоге возникают изменения одного или нескольких оснований, вставки и делеции, образование тиминовых димеров, одно- и двухцепочечные разрывы ДНК, образование сшивок между основаниями одной цепи или комплементарными цепями ДНК, между ДНК и белковыми молекулами. Несмотря на это, в целом геном остается свободным от «ошибок», так как клетка имеет механизмы детекции и репарации поврежденной ДНК. Поврежденное основание может быть восстановлено непосредственно его заменой или обратной химической реакцией (direct repair), в других случаях необходимы более сложные процессы, обеспечивающие удаление поврежденного участка ДНК и достраивание правильной последовательности с использованием комплементарной цепи, редко — гомологичной хромосомы. Такая репарация ДНК — сложный многоступенчатый процесс взаимодействия нескольких каскадных путей. Процесс репарации происходит на разных этапах клеточного цикла. При АФ нарушается способность клетки исправлять определенный тип повреждений ДНК — поперечные межхроматидные сшивки (DNA interstrand crosslink), которые препятствуют работе репликационной вилки. Поперечные межхроматидные сшивки формируются как под воздействием продуктов естественного метаболизма клетки (в первую очередь эндогенных альдегидов, но также и активных форм кислорода), так и под воздействием химических веществ, в частности химиотерапевтических препаратов. К такому типу химических веществ относятся алкилирующие соединения, имеющие в своем составе две активные алкильные группы, обеспечивающие им активное связывание с определенными основаниями: цисплатин, митомицин С, азотистый иприт, псорален, диэпоксибутан, мелфалан, циклофосфамид, мустарген, стрептозоцин. Протеины, функция которых нарушается при АФ, задействованы во всех этапах репарации межхроматидной поперечной сшивки. Этот сложный многоступенчатый процесс получил название FA-pathway, а протеины, задействованные в нем, — АФ-протеины. Ключевую роль в этом процессе играет моноубиквитинирование гена FANCD2, который координирует процессы вырезки поврежденных нуклеотидов, прямое достраивание поврежденного участка и гомологичную рекомбинацию. При АФ клетка не способна адекватно исправлять повреждения ДНК, накопление поломок которой может приводить к костномозговой недостаточности, аномалиям физического развития и предрасположенности к развитию опухолей.
Спектр мутаций, которые приводят к АФ:
1) FANCA.
Мутации в этом гене — самые распространенные и встречаются в 60–70 % случаев АФ. Известно более 100 мутаций, из которых около трети приходится на точечные, еще треть представляют собой микроделеции, и около 40 % представлены крупными делециями. Описаны также и малые дупликации. По действию мутации в гене FANCA могут быть гипоморфными, т. е. приводить к частичной потере функции белка; они характеризуются более мягкими клиническими проявлениями, однако большая часть мутаций вызывает полную потерю функции. Ряд мутаций в гене FANCA имеет повышенную частоту распространения. Так, микроделеция в 38-м экзоне с.3788_3790delTCT — самая распространенная мутация при АФ в мире (20,7 % всех аллелей с мутацией). При этом она встречается у 80 % пациентов с АФ с Канарских островов, где частота встречаемости АФ достигает 1:16000 новорожденных. Кроме того, эта мутация встречается в 51 % случаев АФ в Бразилии. Для подтверждения «эффекта основателя» для данной мутации был проведен анализ гаплотипа пациентов путем изучения вариабельных тандемных повторов и однонуклеотидных полиморфизмов гена FANCA у 28 пациентов с мутацией с.3788_3790delTCT из различных частей света. Все, за исключением одного пациента, имели общего предка. По всей видимости, Канарские острова послужили местом происхождения и распространения заболевания из Европы в Америку, так как несколько веков назад практически все суда из Испании в Америку шли через Канарские острова. Однако, учитывая, что эта мутация тем не менее составляет 2–5 % всех FANCA-мутаций, существует также и мнение, что она связана с явлением существования определенных участков генома с повышенной мутационной способностью, так называемых hotspot. Другой пример «эффекта основателя» — мутация с.295С>Т, которую выявляют почти во всех случаях АФ у испанских цыган. При этом носительство этой мутации среди испанских цыган определено как 1:67. Функционально и фенотипически варианты мутаций в гене FANCA проявляются одинаково.
2) FANCC.
Мутации в гене FANCC встречаются в 10–15 % случаев АФ, почти 90 % случаев представлено двумя мутациями — с.711+4А>Т и delG332. Самая частая мутация в гене FANCC — с. 711+4А>Т, ее выявляют в гомозиготном состоянии в 80 % случаев АФ у евреев-ашкенази. Частота гетерозиготного носительства этой мутации среди евреев-ашкенази достигает 40 %. При этом встречаются и спорадические случаи. Генотип FANCC с.711+4А>Т также распространен среди больных АФ в Японии. В Нидерландах более чем 50 % случаев АФ — это гомозиготные носители мутации с.67delG (также известна как delG332), приводящей к сдвигу рамки считывания в гене FANCC.
3) FANCG.
FANCG задействован в 10 % случаев АФ. Встречаются практически все типы мутаций, за исключением крупных делеций. Клинически характеризуются более частым и быстрым развитием миелодисплазии или лейкоза. Встречаются как спорадические случаи, так и этнически ассоциированный вариант — около 80 % случаев АФ у чернокожих южноафриканцев (Bantu-speakers) (Южная Африка, Свазиленд, Малави и Мозамбик) имеют мутацию 637_643delTACCGCC. Заболевание характеризуется частыми нарушениями пигментации кожи, слабовыраженными аномалиями развития и сравнительно поздними, но тяжелыми гематологическими проявлениями, обусловленными в том числе и поздним обращением к врачу.
Развитие костномозговой недостаточности связывают с повышенным апоптозом гемопоэтических клеток, однако истинные патогенетические механизмы костномозговой недостаточности при АФ мало изучены из-за сложности получения адекватной биологической модели развития заболевания. Последние исследования на ксенографтных моделях и in vitro показали, что в ответ на накопление нерепарированных повреждений ДНК происходит активация р53 проапоптотического пути и запуск поздней р21(Cdkn1a)-зависимой блокировки клеточного цикла в фазе G0/G1 с последующей элиминацией ранних гемопоэтических предшественников из костного мозга. Этот механизм запускается в пренатальном периоде на этапе формирования пула стволовых клеток и ранних клеток-предшественников гемопоэза, что приводит к значительному снижению их количества. Накопление дефектов ДНК после рождения в результате различных физико-химических воздействий усугубляет нарушение гемопоэза. Кроме выраженного апоптоза ранних клеток-предшественников происходит нарушение базовых свойств стволовых кроветворных клеток — способности к самоподдержанию собственной популяции, пролиферации и дифференцировке в различные линии гемопоэза. Генетическая нестабильность при АФ реализуется в повышенной частоте развития ряда опухолей, наиболее частые — ОМЛ и плоскоклеточный рак кожи головы и шеи, слизистых оболочек рта и мочеполового тракта — то есть тканей, характеризующихся высокой пролиферативной активностью.
Клиническая картина
Признаки и симптомы, а также частота их встречаемости указаны в таблице 1.
Таблица 1 | Признаки и симптомы анемии Фанкони
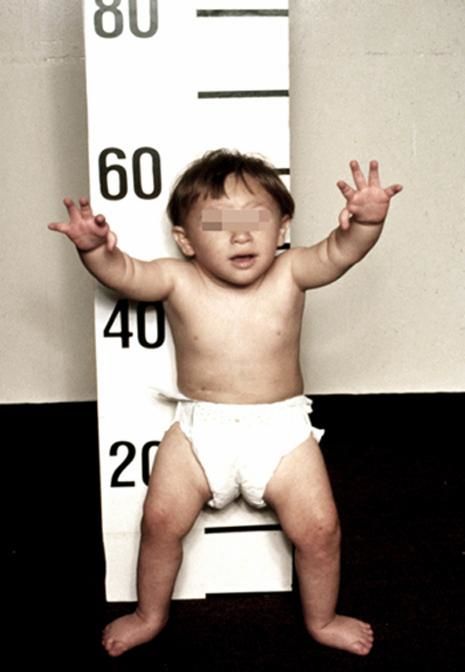
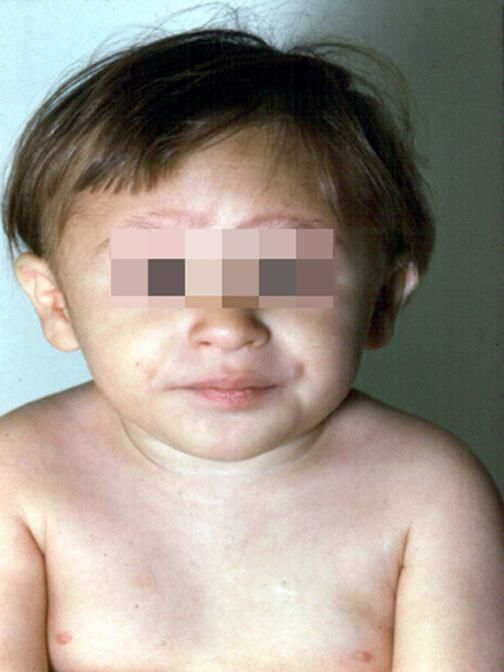
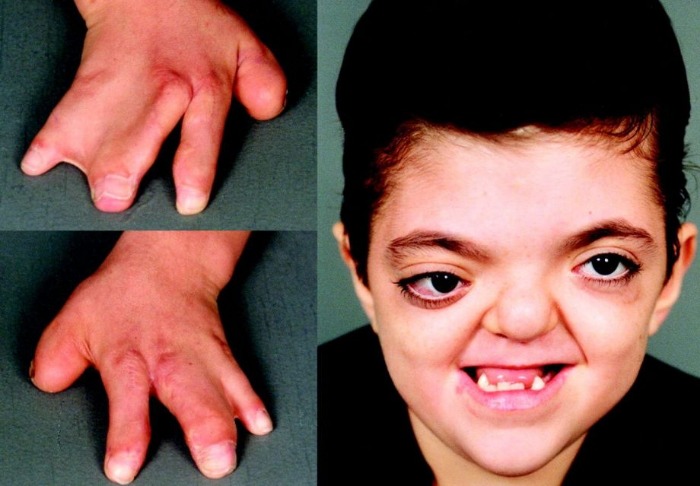
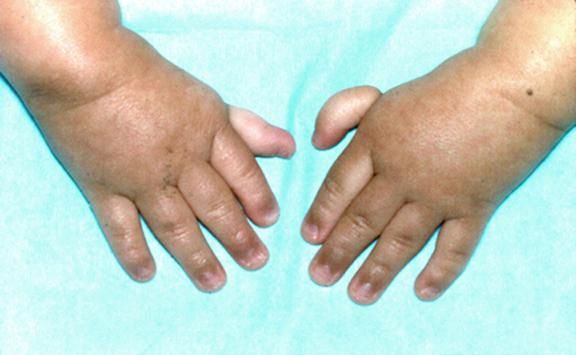
Рисунки 1–5 | Пороки развития, наблюдаемые при анемии Фанкони.
Диагностика
Помимо тщательного физикального осмотра, сбора семейного анамнеза, общего анализа крови и пункции костного мозга для более точной диагностики используются такие методы лабораторной диагностики, как тест на ломкость хромосом, метод MLPA, высокопроизводительное секвенирование и секвенирование по Сэнгеру.
1) Тест на ломкость хромосом.
«Золотым стандартом» скрининга для выявления АФ был и остается тест с диэпоксибутаном (1,3-butadienediepoxide) и его вариант с митомицином С (MMC). Еще в начале изучения АФ было отмечено, что фибробласты и лимфоциты больных АФ в культуре клеток демонстрируют спонтанную повышенную ломкость хромосом. Значительные различия в уровне спонтанных аберраций у больных (вплоть до отсутствия таковых) требовали унифицированного и точного метода детекции. Позже была показана повышенная чувствительность клеток больных АФ к действию алкилирующих агентов, вызывающих поперечные сшивки между нуклеотидами, что препятствует образованию нормальной репликативной вилки для запуска процесса репарации ДНК, позже получивших общее название interstrand cross-link agent. На основании этого был предложен цитогенетический метод диагностики АФ: после обработки лимфоцитов или фибробластов алкилирующим веществом (в нелетальной для клеток концентрации) определяют частоту и спектр спонтанных и индуцированных in vitro хромосомных аберраций. Обычно ставится несколько параллельных клеточных культур, стимулированных фитогемагглютинином лимфоцитов периферической крови: без добавления алкилирующего агента (для определения спонтанного уровня аберраций) и с его добавлением в разной концентрации. Затем в метафазных пластинках подсчитывают число хромосомных разрывов. Для АФ характерны разрывы хромосом с образованием радиальных фигур, фрагментов, хромосомных и хроматидных разрывов, а также три- и тетрарадикалов. При анализе результатов учитывают число разрывов по отношению к числу проанализированных метафаз, процент клеток с разрывами и ряд других показателей. Необходимо отметить, что значения, при которых тест на ломкость хромосом считается положительным, в различных лабораториях варьируются. Тест не имеет 100 % специфичности. Положительный тест на ломкость хромосом бывает у пациентов с синдромом Ниймеген (мутации в гене NBS1), синдромом Робертса (мутации в гене ESCO2), Warsaw breakage syndrome (мутации в гене DDX11), синдромом Блюма (ген BLM), врожденном дискератозе и некоторых других синдромах. В то же время АФ-подобные синдромы могут оказаться действительно АФ с соответствующим генетическим дефектом.
2) Секвенирование по Сэнгеру долгие годы было основным методом определения мутаций при АФ. Учитывая значительные размеры генов, секвенирование каждого из них представляет собой довольно трудоемкий и дорогостоящий процесс. Его обычно проводили после анализа на группу комплементации, предварительно определив вероятный ген. Анализ нуклеотидной последовательности для всех известных АФ-генов затруднен количеством возможных мутаций в каждом, их разнообразием, в том числе в виде крупных инсерций или делеций (indel-мутации). Их длина может варьировать от одного до нескольких сотен и даже тысяч нуклеотидных оснований, что подразумевает использование совершенно разных методов молекулярно-генетического исследования.
3) Метод MLPA (мультиплексная амплификация лигазносвязанных проб) предназначен для определения делеций и амплификаций определенных последовательностей гена длиной до нескольких десятков нуклеотидов. Одновременно может быть исследовано до 60 таких последовательностей, что позволяет выявить как сравнительно небольшие делеции, так и делеции отдельных экзонов и целого гена. Метод MLPA используют для инициального скрининга делеции в гене FANCA, параллельно ген FANCA полностью секвенируют. Если пациент мужского пола, его исследуют на наличие делеций ген FANCB. Выявленные делеции желательно подтвердить другим методом. При этом методы, которые могут быть использованы, требуют индивидуальной разработки в каждом конкретном случае: количественная ПЦР, ПЦР длинных фрагментов (long range PCR) и хромосомный микроматричный анализ.
4) Высокопроизводительное секвенирование.
Метод, который позволяет одномоментно анализировать от нескольких генов до полного генома, является наиболее подходящим для определения мутаций при АФ. Возможны несколько вариантов теста. Первый — секвенирование экзома, позволяет получить максимальный объем информации. Второй — секвенирование ограниченного числа интересующих и уже описанных в литературе генов (таргетное ресеквенирование), список которых можно дополнять или моделировать в соответствии с потребностями исследования. Современные коммерческие панели генов, как правило, помимо генов АФ включают большое число генов, ответственных за развитие других врожденных синдромов, в том числе и АФ-подобных. Внедрение в практику высокопроизводительного секвенирования позволяет избежать последовательного трудоемкого исследования каждого из известных генов методом секвенирования по Сэнгеру, однако пока не позволяет с должной уверенностью выявить крупные делеции и дупликации. Найденные мутации требуют подтверждения одним из других подходящих методов. При обнаружении новых мутаций необходимо подтверждение их патогенности в функциональном тесте и др.
Пренатальная и преимплантационная диагностика должна проводиться в первую очередь в семьях, где ранее были установлены патогенетические мутации. В этом случае проводится целенаправленный поиск известной мутации. Материалом для диагностики служат клетки плода, получаемые путем биопсии ворсин хориона на 10–12 неделе беременности. Следует помнить, что генетический анализ занимает не менее 2–3 недель. Если нет возможности провести молекулярно-генетическое исследование, возможно выполнение теста на ломкость хромосом клеток ворсин хориона на 10–12 неделе беременности либо при амниоцентезе на 15–18 неделе. Однако молекулярно-генетическое исследование предпочтительнее.
Лечение
Основной метод лечения АФ — аллогенная трансплантация гемопоэтических стволовых клеток.
Из медикаментозного лечения в настоящий момент применяют андрогены. Препараты этой группы (оксиметалон, метандростенолон) позволяют достичь гематологического ответа примерно у 50 % больных. Также применение андрогенов значимо увеличивает продолжительность жизни у ответивших на лечение пациентов: медиана продолжительности жизни составляет 9 лет после установления диагноза против 2,5 лет соответственно для тех пациентов, у которых лечение андрогенами не было эффективным.
При возникновении сопутствующих заболеваний, например, инфекций, обосновано применение гранулоцитарного колониестимулирующего фактора, который способен временно увеличить количество нейтрофилов и облегчить протекание заболевания.
В 2015 году коллективу ученых удалось успешно применить технологию Crispr/Cas9 для редактирования мутации с.67delG гена FANCC.
Источники:
- https://www.bloodjournal.org/content/126/23/3622?sso-checked=true
- https://www.ncbi.nlm.nih.gov/books/NBK1401/
- https://rarediseases.info.nih.gov/diseases/6425/fanconi-anemia
- https://rarediseases.org/rare-diseases/fanconi-anemia/
- https://cyberleninka.ru/article/v/geneticheskaya-diagnostika-anemii-fankoni-obzor-literatury
Источник