Гемоглобин образование разрушение и выведение

Разрушение гемоглобина. Разновидности анемий
При разрыве эритроцитов их гемоглобин почти сразу же фагоцитируется макрофагами во многих частях тела, но особенно клетками Купфера печени и макрофагами селезенки и костного мозга. В течение нескольких следующих часов или дней макрофаги освобождают железо из гемоглобина, и оно возвращается в кровь и переносится трансферрином либо в костный мозг для формирования новых красных клеток крови, либо в печень и другие ткани для хранения в форме ферритина.
Порфириновая часть молекулы гемоглобина превращается макрофагами через ряд стадий в желчный пигмент билирубин, который выделяется в кровь и позднее удаляется из организма путем секреции печенью в желчь.
Анемии
Анемия означает недостаток гемоглобина в крови, причиной может быть либо слишком малое число красных клеток крови, либо слишком малое количество гемоглобина в этих клетках. Далее представлены некоторые типы анемий и их физиологические причины.
а) Анемия, связанная с потерей крови. После острой кровопотери организм возмещает жидкую часть плазмы в течение 1-3 сут, но при этом концентрация красных клеток крови остается низкой. Концентрация эритроцитов обычно восстанавливается до нормы в течение 3-6 нед, если не происходит повторного кровотечения.
Часто при хронической кровопотере у человека железо из кишечника не может всасываться достаточно быстро, чтобы обеспечить адекватное возмещение теряемого с кровью гемоглобина. Формируемые в этом случае красные клетки крови гораздо мельче нормальных эритроцитов и содержат слишком мало гемоглобина, что характерно для микроцитарной гипохромной анемии; такие эритроциты показаны на рисунке.
б) Апластическая анемия. Аплазия костного мозга означает потерю функционирующего костного мозга. Например, у человека, подвергшегося облучению гамма-лучами при взрыве атомной бомбы, может произойти полное разрушение костного мозга с последующим развитием в течение нескольких недель летальной анемии. Тот же эффект могут вызвать избыточная рентгенотерапия, некоторые промышленные химикаты и даже лекарства, к которым у человека может быть повышенная чувствительность.
в) Мегалобластная анемия. На основании изложенного ранее обсуждения роли витамина B12, фолиевой кислоты и внутреннего фактора, секретируемого слизистой желудка, легко понять, что недостаток любого из этих веществ может привести к замедлению репродукции эритроцитов в костном мозге. В результате формируются слишком крупные красные клетки крови разнообразной формы, которые называют мегалобластами.
Следовательно, атрофия слизистой желудка, например при пернициозной анемии, или потеря всего желудка после хирургической тотальной гастрэктомии могут привести к мегалобластной анемии. Мегалобластная анемия часто развивается также у больных с кишечной спру, при которой плохо всасываются фолиевая кислота, витамин B12 и другие соединения витаминов группы В. Поскольку при этих состояниях эритробласты не могут пролиферировать достаточно быстро, чтобы формировать нормальное количество красных клеток крови, те эритроциты, которые формируются, по большей части увеличены в размерах, имеют неправильную форму и ломкие мембраны. Эти клетки легко рвутся, оставляя человека без необходимого количества красных клеток крови.
г) Гемолитическая анемия. Различные аномалии красных клеток крови, многие из которых — наследственные, делают клетки столь хрупкими, что они легко разрываются, проходя через капилляры, особенно в селезенке. Даже если количество формируемых красных клеток крови в норме или значительно ее превышает, как при некоторых гемолитических болезнях, срок жизни хрупкого эритроцита так короток, что клетки разрушаются быстрее, чем могут формироваться нормальные эритроциты; результатом этого является тяжелая анемия. Далее указаны некоторые из таких типов анемий.
При наследственном сфероцитозе красные клетки крови очень мелкие и сферические, а не двояковогнутые диски. Эти клетки не могут выдерживать сдавливания, поскольку не имеют нормальной свободной, мешкообразной клеточной мембраны, характерной для двояковогнутых дисков. При прохождении через пульпу селезенки и некоторые другие сосудистые ложа они легко ломаются даже при небольшом сдавливании.
При серповидно-клеточной анемии, которой болеют 0,3-1,0% коренных жителей Западной Африки, красные клетки крови содержат аномальный гемоглобин — гемоглобин S с поврежденными цепочками в его молекуле. Под действием низких концентраций кислорода такой гемоглобин осаждается в виде длинных кристаллов внутри эритроцита. Эти кристаллы удлиняют клетку и придают ей вид серпа, а не двояковогнутого диска.
Кроме того, осажденный гемоглобин повреждает клеточную мембрану, в результате клетка становится очень хрупкой, что сопровождается тяжелой анемией. Такие больные часто переживают порочный круг событий, называемый кризисом серповидно-клеточной болезни, при котором низкое напряжение кислорода в тканях вызывает образование серповидных форм эритроцитов, что ведет к разрушению красных клеток крови, а значит — к дальнейшему снижению напряжения кислорода, усилению образования серповидных форм и разрушению красных клеток крови. Сразу после начала процесс быстро прогрессирует, приводя в течение нескольких часов к резкому снижению числа красных клеток крови и часто — к смерти.
При гемолитической болезни новорожденных (эритробластозе) антитела от резус-отрицательной (Rh-) матери атакуют резус-положительные (Rh+) эритроциты плода. В результате резус-положительные клетки становятся ломкими, что ведет к их быстрому разрушению, способствуя развитию у новорожденного тяжелой анемии. Чрезвычайно быстрое формирование новых эритроцитов для возмещения разрушенных при гемолитической болезни новорожденных ведет к выделению в кровь из костного мозга большого количества молодых бластных форм красных клеток крови.
– Также рекомендуем “Влияние анемии на кровообращение. Полицитемия – эритремия”
Оглавление темы “Эритропоэз. Белые клетки крови”:
1. Влияние эритропоэтина на эритрогенез. Витамин В12 и фолиевая кислота в эритропоэзе
2. Пернициозная анемия. Образование гемоглобина
3. Связывание гемоглобина с кислородом. Обмен железа
4. Всасывание железа в кишечнике. Длительность жизни эритроцитов
5. Разрушение гемоглобина. Разновидности анемий
6. Влияние анемии на кровообращение. Полицитемия – эритремия
7. Влияние полицитемии на кровообращение. Лейкоциты – белые клетки крови
8. Типы белых клеток крови. Происхождение белых клеток крови
9. Длительность жизни белых клеток крови. Нейтрофилы и макрофаги
10. Фагоцитоз. Механизмы и значение фагоцитоза
За сутки у человека распадается около 9 г гемопротеинов, в основном это гемоглобин эритроцитов.
Эритроциты в норме живут 90-120 дней, после чего лизируются в клетках ретикулоэндотелиальной системы – макрофагах селезенки (главным образом), купферовских клетках печени и макрофагах костного мозга. При разрушении эритроцитов в кровеносном русле высвобождаемый гемоглобин образует комплекс с белком-переносчиком гаптоглобином (фракция α2-глобулинов крови) и также переносится в клетки РЭС селезенки, печени и костного мозга.
Синтез билирубина
В клетках РЭС гем в составе гемоглобина окисляется молекулярным кислородом. В реакциях последовательно происходит разрыв метинового мостика между 1-м и 2-м пиррольными кольцами гема с их восстановлением, отщеплением железа и белковой части и образованием оранжевого пигмента билирубина. Высвобождаемое железо может либо запасаться в клетке в комплексе с ферритином, либо выделяться наружу и связываться с трансферрином.
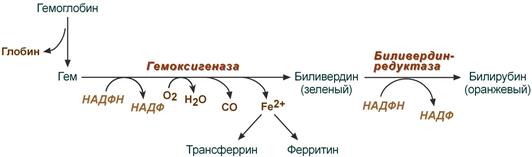
Реакции распада гемоглобина и образования билирубина
Билирубин – токсичное, жирорастворимое вещество, способное разобщать окислительное фосфорилирование в клетках. Особенно чувствительны к нему клетки нервной ткани.
Строение билирубина
Выведение билирубина
Из клеток ретикуло-эндотелиальной системы билирубин попадает в кровь. Здесь он находится в комплексе с альбумином плазмы, в гораздо меньшем количестве – в комплексах с металлами, аминокислотами, пептидами и другими малыми молекулами. Образование таких комплексов не позволяет выделяться билирубину с мочой. Билирубин в комплексе с альбумином называется свободный (неконъюгированный) или непрямой билирубин.
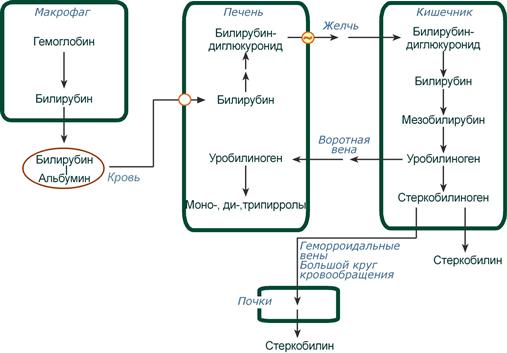
Этапы метаболизма билирубина в организме
Из сосудистого русла в гепатоциты билирубин попадает с помощью белка-переносчика (транспортный белок органических анионов) или по механизму флип-флоп. Далее при участии цитозольного связывающего белка лигандина (Y-протеин) билирубин транспортируется в ЭПР, где протекает реакция связывания билирубина с УДФ-глюкуроновой кислотой, при этом образуются моно- и диглюкурониды. Кроме глюкуроновой кислоты, в реакцию конъюгации могут вступать сульфаты, фосфаты, глюкозиды.
Билирубин-глюкуронид получил название связанный (конъюгированный) или прямой билирубин.
После образования билирубин-глюкурониды АТФ-зависимым переносчиком секретируются в желчные протоки и далее в кишечник, где при участии бактериальной β-глюкуронидазы превращаются в свободный билирубин. Одновременно, даже в норме (особенно у взрослых), некоторое количество билирубин-глюкуронидов может попадать из желчи в кровь по межклеточным щелям.
Таким образом, в плазме крови обычно присутствуют две формы билирубина: свободный (непрямой), попадающий сюда из клеток РЭС (80% и более всего количества), и связанный (прямой), попадающий из желчных протоков (в норме не более 20%).
Термины “связанный“, “конъюгированный“, “свободный“, “несвязанный” отражают взаимодействие билирубина и глюкуроновой кислоты (но не билирубина и альбумина!).
Термины “прямой” и “непрямой” введены, исходя из возможности химической реакции билирубина с диазореактивом Эрлиха. Связанный билирубин реагирует с реактивом напрямую, без добавления дополнительных реагентов, т.к. является водорастворимым. Несвязанный (жирорастворимый) билирубин требует добавочных реактивов, реагирует не прямо.
Превращение в кишечнике
В кишечнике билирубин подвергается восстановлению под действием микрофлоры до мезобилирубина и мезобилиногена (уробилиногена). Часть уробилиногена всасывается и с кровью портальной вены попадает в печень, где либо распадается до моно-, ди- и трипирролов, либо окисляется до билирубина и снова экскретируется. При этом при здоровой печени в общий круг кровообращения и в мочу мезобилирубин и уробилиноген не попадают, а полностью задерживаются гепатоцитами.
Оставшаяся в кишечнике часть пигментов ферментами бактериальной флоры толстого кишечника восстанавливается до стеркобилиногена. Далее
- малая часть стеркобилиногена может всасываться и катаболизировать в печени, подобно уробилиногену,
- незначительное количество стеркобилиногена через геморроидальные вены попадает в большой круг кровообращения, отсюда в почки и в мочу. После окисления на воздухе из стеркобилиногена образуется стеркобилин мочи,
- однако основное количество стеркобилиногена достигает нижних отделов толстого кишечника и выделяется. В прямой кишке и на воздухе стеркобилиноген окисляется в стеркобилин, окрашивая кал,
- аналогично уробилиноген, появляющийся в моче при патологии печени, окисляется в уробилин.
Очень часто стеркобилиноген, содержащийся в нормальной моче, называют уробилиногеном. И в клинической практике обычно не проводят различий между стеркобилиногеном и уробилиногеном мочи, их рассматривают как один пигмент – урохромы (уробилиноиды), что может создавать некоторую путаницу при оценке результатов анализа.
Пернициозная анемия. Образование гемоглобинаНарушение созревания, связанное с недостаточным всасыванием витамина В12 из желудочно-кишечного тракта: а) Пернициозная анемия. Распространенной причиной нарушения созревания красных клеток крови является недостаточное всасывание витамина В12 из желудочно-кишечного тракта. Это часто происходит при заболевании пернициозной анемией, при которой основной патологией является атрофический гастрит, нарушающий нормальную секреторную функцию желудка. Париетальные клетки желудочных желез секретируют гликопротеин, называемый внутренним фактором, который соединяется с витамином В12 пищи и делает этот витамин доступным для всасывания в кишечнике. Это происходит следующим образом: (1) внутренний фактор прочно связывается с витамином B12. В таком связанном состоянии витамин В12 защищается от переваривания желудочно-кишечными секретами; (2) находясь в связанном состоянии, внутренний фактор связывается со специфическими рецепторами на мембранах клеток щеточной каемки слизистой подвздошной кишки; (3) затем в течение следующих нескольких часов витамин В12 транспортируется в кровь путем пиноцитоза; в результате внутренний фактор и витамин вместе переносятся через мембрану. Следовательно, недостаток внутреннего фактора ведет к уменьшению доступности витамина В12 из-за нарушения всасывания витамина. Сразу после всасывания из желудочно-кишечного тракта витамин B12 сначала накапливается в печени в большом количестве, затем медленно выделяется по мере потребности в нем костного мозга. Минимальное количество витамина B12, необходимое ежедневно для поддержания нормального созревания красных клеток крови, составляет всего 1-3 мкг, а нормальное накопление в печени и других тканях тела примерно в 1000 раз больше этого количества. Следовательно, обычно для развития анемии в связи с нарушением созревания эритроцитов недостаточность всасывания В12 должна продолжаться 3-4 года. б) Недостаточность созревания, связанная с дефицитом фолиевой кислоты (птероилглутаминовой кислоты). Фолиевая кислота является нормальным компонентом овощей, некоторых фруктов и мяса (особенно, печени). Однако она легко разрушается во время приготовления пищи. Кроме того, люди с нарушением всасывания в желудочно-кишечном тракте, например при часто встречающемся заболевании тонкого кишечника, которое называют спру, обычно имеют серьезные затруднения всасывания и фолиевой кислоты, и витамина B12. Следовательно, во многих случаях причиной нарушений созревания является недостаточность всасывания в кишечнике и фолиевой кислоты, и витамина B12. Формирование гемоглобинаСинтез гемоглобина начинается в проэритробластах и продолжается даже на стадии ретикулоцита красных клеток крови. Следовательно, когда ретикулоциты оставляют костный мозг и проходят в кровоток, они продолжают формировать минимальные количества гемоглобина в течение примерно следующего дня до тех пор, пока не станут зрелыми эритроцитами.
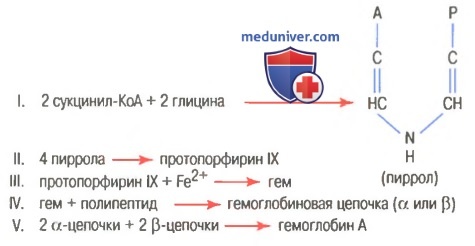
На рисунке выше показаны основные химические этапы формирования гемоглобина. Сначала сукцинил-КоА, формируемый в метаболическом цикле Кребса, связывается с глицином, образуя молекулу пиррола. В свою очередь, четыре молекулы пиррола объединяются, образуя протопорфирин IX, который затем соединяется с железом, формируя молекулу гема. Наконец, каждая молекула гема присоединяется к синтезируемому рибосомами длинному полипептиду глобину, формируя субъединицу гемоглобина, называемую гемоглобиновой цепочкой (для облегчения понимания просим вас изучить рисунок ниже). Каждая цепочка имеет молекулярную массу около 16000; в свою очередь, четыре такие цепочки свободно связываются вместе, формируя целую гемоглобиновую молекулу.
Существуют несколько легких вариаций гемоглобиновых цепочек в зависимости от аминокислотного состава полипептидной части субъединицы. Разные типы цепочек обозначают как альфа, бета, гамма и дельта. Наиболее распространенная форма гемоглобина взрослого человека — гемоглобин А. Он представляет собой комбинацию двух альфа-цепочек и двух бета-цепочек. Гемоглобин А имеет молекулярную массу 64458. Поскольку каждая гемоглобиновая цепочка имеет простетическую группу в виде гема, содержащего атом железа, ясно, что каждая молекула гемоглобина, состоящая из четырех таких цепочек, содержит четыре атома железа. Каждый из них может свободно связываться с одной молекулой кислорода, обеспечивая возможность транспортировки каждой молекулой гемоглобина четырех молекул (или восьми атомов) кислорода. Типы гемоглобиновых цепочек в молекуле гемоглобина определяют сродство гемоглобина к кислороду. Патологические изменения цепочек могут менять физические характеристики гемоглобиновой молекулы. Например, при серповидно-клеточной анемии в одном участке каждой из двух бета-цепочек аминокислота валин заменяется глутаминовой кислотой. При низком парциальном давлении кислорода этот тип гемоглобина формирует внутри эритроцитов вытянутые кристаллы, длина которых иногда достигает 15 мкм. Они делают практически невозможным прохождение эритроцитов через многие мелкие капилляры, и острые концы кристаллов, вероятно, разрывают клеточные мембраны, способствуя развитию серповидно-клеточной анемии. – Также рекомендуем “Связывание гемоглобина с кислородом. Обмен железа” Оглавление темы “Эритропоэз. Белые клетки крови”: |
Гемоглобинопатия – это наследственные заболевания с единой проблемой – образованием аномальной формы гемоглобина, например, серповидноклеточная анемия S и талассемия.
Гемоглобинопатии носят эндемический характер – они возникают в определенном географическом районе, например, в Средиземноморье, Африке, Юго–Восточной Азии. В нашей стране они тоже встречаются.
Что такое гемоглобинопатия
Гемоглобинопатии – это заболевания, вызванные выработкой и присутствием аномальной формы гемоглобина.
Гемоглобин состоит из гема (частей, содержащих железо) и глобина (частей белка, состоящих из аминокислотных цепей). Молекулы гемоглобина (Hb или Hgb) находятся в красных кровяных тельцах. Их задача – связывать кислород в легких и передавать его тканям и органам, где они его выделяют.
Строение гемоглобина
Существует несколько типов цепей глобина: альфа, бета, дельта и гамма.
Типы нормального гемоглобина:
- A – HbA: составляет около 95-98% от общего гемоглобина у взрослых людей. Он содержит 2 альфа (α) цепи и две бета (β) цепи.
- A2 – HbA2: составляет около 2-3% от общего гемоглобина. Он содержит 2 цепи альфа (α) и две цепи дельта (δ).
- F (HbF): составляет около 2% от общего гемоглобина взрослого человека. Он содержит 2 альфа (α) цепи и две гамма (γ) цепи. Этот гемоглобин в основном вырабатывается у плода, его производство значительно снижается вскоре после рождения и достигает уровня взрослого человека в течение 1-2 лет.
К гемоглобинопатиям относятся: структурные варианты гемоглобина, гемоглобин S, серповидноклеточная анемия, гемоглобинопатия C, гемоглобинопатия E, талассемия, гемоглобин Бартс, наследственная персистенция гемоглобина плода.
Причины развития гемоглобинопатии
Гемоглобинопатии возникают в случае генетических изменений генов глобина, которые приводят к изменению аминокислот, составляющих белок глобина. Эти изменения влияют на:
- структуру гемоглобина, например, гемоглобин S, который вызывает серповидно-клеточную анемию;
- его поведение;
- количество продуцируемого вещества (талассемия);
- стабильность.
Серповидно-клеточная анемия
Существует четыре гена, кодирующих цепь альфа-глобина, и два гена, кодирующих цепь бета-глобина. Наиболее частым заболеванием, связанным с изменением альфа-цепи, является альфа-талассемия. Его тяжесть зависит от количества пораженных генов.
Талассемия характеризуется снижением продукции одной из цепей глобина, дисбалансом между альфа- и бета-цепями в гемоглобине A (альфа-талассемия) или увеличением малых форм, таких как Hb A2 или Hb F (бета-талассемия).
Изменения бета-цепей гемоглобина являются врожденными, аутосомно-рецессивными. Это означает, что больной человек должен иметь две дефектные копии гена, каждая от одного из родителей. Если один ген нормален, а другой дефектен, человек гетерозиготен, и мы называем его носителем. Аномальный ген может быть передан любому из потомков. Если рассматриваемый человек является гетерозиготным носителем, он может не иметь никаких симптомов и носительство не влияет на его здоровье.
Если происходят две модификации одного и того же бета-гена, человек гомозиготен по этому гену. Его организм может производить дефектный гемоглобин – возникает гемоглобинопатия с симптомами и потенциальными осложнениями. Степень тяжести зависит от генетической мутации и варьируется от случая к случаю. Копию гена можно передать потомству.
Если два аномальных бета-гена являются врожденными, человек является двойным, смешанным гетерозиготным. У него будут симптомы одной или обеих гемоглобинопатий. Один из аномальных бета-генов будет передаваться каждому из потомков.
Были идентифицированы сотни гемоглобинопатий в бета-цепях. Хотя лишь некоторые из них являются общими и клинически значимыми.
Клинические признаки и симптомы
Признаки и симптомы различаются по типу гемоглобинопатии и возможному сочетанию нескольких гемоглобинопатий. Некоторые приводят к усилению распада эритроцитов (гемолизу), уменьшению их общего количества и развитию анемии.
Клинические признаки включают:
- слабость, утомляемость;
- недостаток энергии;
- желтуха;
- бледность кожи.
Утомляемость
К серьезным клиническим признакам относятся:
- приступы сильной боли;
- удушье;
- увеличение селезенки;
- нарушения роста у детей;
- боль в верхней части живота (вызванная желчными камнями).
Удушье
Общие гемоглобинопатии
Красные кровяные тельца, содержащие аномальный гемоглобин, могут не переносить кислород достаточно эффективно. Они могут разрушаться раньше (чем в здоровых клетках крови) и развиваться гемолитическая анемия. Выявлены сотни гемоглобинопатий, но лишь некоторые из них являются общими и клинически значимыми.
Одной из наиболее распространенных гемоглобинопатий является серповидно-клеточная анемия с присутствием гемоглобина S. Это приводит к изменению формы – серповидно-клеточной – эритроцитов и снижению их выживаемости. Гемоглобин С может вызвать легкую гемолитическую анемию. Гемоглобин E обычно не приводит к развитию каких-либо или только очень легких клинических симптомов.
- Талассемия: самая распространенная генетическая аномалия в мире. Она часто встречается в Средиземноморье, на Ближнем Востоке и в Юго-Восточной Азии. Более легкая форма талассемии также встречается, например, у людей, родившихся в Чехии.
- Гемоглобин S: это основной гемоглобин людей с серповидно-клеточной анемией. В среднем эта мутация есть в одном из двух бета-генов у 8% американцев и африканцев. Возникновение этих мутаций в наших широтах встречаеся редко. Пациенты с заболеванием HbS имеют две аномальные цепи бета (b s) и две нормальные цепи альфа (a). Когда эритроциты, содержащие гемоглобин S, подвергаются действию пониженного количества кислорода (как это может быть в случае повышенной физической нагрузки или инфекционного заболевания легких), они деформируются, принимая форму полумесяца. Серповидные эритроциты могут блокировать периферические кровеносные сосуды и вызывать нарушения кровотока и боль. У них пониженная способность переносить кислород и более короткий срок жизни. Одна копия б не вызывает клинических проявлений, если не сочетается с другой мутацией гемоглобина, такой как HbC (b C) или бета-талассемия.
- Гемоглобин C: около 25% жителей Западной Африки и 2-3% афроамериканцев гетерозиготны по гемоглобину C (у них есть одна копия B C). Но заболевают только гомозиготные люди с обоими дефектными генами (b C). Обычные симптомы – легкая гемолитическая анемия с небольшим или средним увеличением селезенки.
- Гемоглобин E: вторая по распространенности гемоглобинопатия в мире с изменением бета-цепей. Патология очень часто встречается в Юго-Восточной Азии, особенно в Камбодже, Лаосе и Таиланде, а также частично в Северо-Восточной Азии. Есть случаи и в нашей стране. Люди с гомозиготным Hb E (две копии b E) обычно имеют легкую гемолитическую анемию, микроциты (маленькие красные кровяные тельца) и слегка увеличенную селезенку. Одна копия гемоглобина E не вызывает клинических признаков, если не сочетается с другой мутацией, такой как одна из бета-талассемии.
Талассемия
Необычные гемоглобинопатии
Существует ряд гемоглобинопатий, некоторые из которых не проявляются – они не вызывают никаких клинических признаков или симптомов. Другие, в свою очередь, влияют на функциональность и / или стабильность молекулы гемоглобина. Примерами являются гемоглобин D, гемоглобин G, гемоглобин J, гемоглобин M и гемоглобин Constant Spring. Мутации в гене альфа-цепи глобина приводят к образованию аномально длинных альфа (а) цепей, которые вызывают нестабильность в молекуле гемоглобина.
Другие примеры мутаций бета-цепи:
- Гемоглобин F: Hb F в основном вырабатывается в организме будущего ребенка (плода), и его функция заключается в переносе кислорода в среде с низким содержанием кислорода. Продукция гемоглобина F снижается сразу после рождения и стабилизируется на уровне взрослого человека до 1-2 лет. Гемоглобин F может быть повышен при некоторых врожденных заболеваниях. При бета-талассемии его уровень может быть нормальным или повышенным, но часто повышен при серповидно-клеточной анемии и сочетании серповидно-клеточной анемии с бета-талассемией. Пациенты с серповидно-клеточной анемией и повышенным Hb F часто имеют более легкое течение болезни, поскольку Hb F предотвращает серповидное движение красных кровяных телец. Уровни Hb F повышены в редком состоянии, называемом врожденным постоянством выработки гемоглобина плода (HPFH). Люди с повышенным уровнем гемоглобина F не имеют клинических признаков. HPFH вызывается разными генными мутациями у разных этнических групп. Hb F также может быть повышен при некоторых приобретенных состояниях, влияющих на выработку красных кровяных телец. Например, лейкемия и миелопролиферативные заболевания часто сопровождаются небольшим повышением уровня гемоглобина F.
- Гемоглобин H: HbH – это аномальный гемоглобин, который возникает в некоторых случаях альфа-талассемии. Его образование является ответом на фундаментальный недостаток альфа (а) цепей. Hb H состоит из четырех цепей бета (b) глобина. Хотя каждая из цепей бета-глобина нормальна, комплекс из четырех цепей бета нормально не функционирует. Обладает повышенным сродством к кислороду, плохо выделяет кислород клеткам тканей. Присутствие гемоглобина H также связано со значительным распадом эритроцитов (гемолизом), который возникает в результате осаждения нестабильного гемоглобина внутри красных кровяных телец.
- Hemoglobin Barts: Hb Barts вырабатывается в организме будущего ребенка с альфа-талассемией при условии, что все четыре гена, отвечающие за производство гемоглобина альфа, отсутствуют. Таким образом, не может образовываться гемоглобин HbA, HbA 2 и HbF. Гемоглобин Бартс состоит из четырех гамма (g) цепей и имеет высокое сродство к кислороду. Это состояние несовместимо с жизнью и обычно приводит к внутриутробной гибели плода.
Некоторые люди могут унаследовать два гена с разными мутациями, каждый от одного из родителей. Таких людей называют двойными или смешанными гетерозиготами.
Обследование: лабораторные тесты
Исследование на гемоглобинопатию проводится в следующих случаях:
- Выявление гемоглобинопатий у бессимптомных родителей больных детей.
- Выявление гемоглобинопатий у пациента с необъяснимой анемией, микроцитозом и / или гипохромией. Анализ может быть выполнен как часть теста на анемию.
- Скрининг на гемоглобинопатии у новорожденных – только в США и некоторых регионах с повышенной заболеваемостью.
- Пренатальный скрининг проводится в некоторых регионах с высокой частотой гемоглобинопатий (особенно в Африке).
На результаты тестов на гемоглобинопатию может повлиять переливание крови. Поэтому после переливания крови, прежде чем сдать анализ, пациенту следует подождать несколько месяцев. Тем не менее пациентам с серповидно-клеточной анемией после переливания крови рекомендуется сдать анализ крови, чтобы увидеть, достаточно ли гемоглобина в крови, и снизить риск повреждения организма серповидными эритроцитами.
Обследование гемоглобинопатий основано на обнаружении и оценке «нормальности» эритроцитов и гемоглобина в эритроцитах, а также на исследовании конкретной мутации гена. Каждый из тестов является частью головоломки, предоставляющей важную информацию о том, какая гемоглобинопатия присутствует. Для проверки гемоглобинопатии используются следующие тесты:
- Анализ крови. Анализ крови дает быструю информацию о клетках, циркулирующих в крови. Помимо прочего, результаты анализа крови показывают, сколько красных кровяных телец (эритроцитов) содержится в крови, какого они размера и формы, а также сколько там гемоглобина. Размер эритроцита определяет средний объем эритроцитов (MCV). Обнаружение пониженного MCV (микроцитоз, наличие небольших эритроцитов) часто сначала указывает на возникновение талассемии. Если MCV низкий и дефицит железа исключен, пациенты могут быть носителями талассемии или гемоглобинопатии, которые также вызывают микроцитоз (например, HbE).
- Анализ ДНК. Этот анализ используется для скрининга мутаций и делеций в альфа- и бета-областях глобиновых генов. Иногда обследуются все члены семьи. Задача в том, чтобы определить конкретный тип мутации, встречающейся в семье, и выявить всех носителей. ДНК-тесты не являются обычным тестом, но они могут помочь диагностировать гемоглобинопатию и выявить носителей.
- Мазок периферической крови (микроскопический дифференциальный подсчет лейкоцитов, считываемый по мазку периферической крови). Тест проводится путем формирования тонкого слоя крови на предметном стекле и окрашивания его специальными красителями. Образец крови, обработанный таким образом, затем оценивается лаборантом под микроскопом. Специалист определяет количество и тип белых и красных кровяных телец и тромбоцитов. Оценивает, являются ли они нормальными и зрелыми.
Анализ крови
При гемоглобинопатии эритроциты могут быть в следующих формах:
- Микроциты (меньше нормального).
- Гипохромные (более бледные, с пониженным гемоглобином).
- Разных размеров (анизоцитоз) и формы (пойкилоцитоз, например, серповидно-клеточные клетки).
- С ядром (в незрелых эритроцитах) или с включениями.
- С неравномерным распределением гемоглобина (клетки-мишени, которые под микроскопом выглядят как «бычий глаз»).
Наличие более высокого процента аномально выглядящих эритроцитов означает более высокую вероятность наличия заболевания.
С помощью тестов на гемоглобинопатию и их комбинаций можно диагностировать наиболее распространенные гемоглобинопатии. Эти тесты могут помочь выявить пациентов с сочетанием различных гемоглобинопатий (смешанные гетерозиготы).
Лечение гемоглобинопатии
В настоящее время гемоглобинопатии – неизлечимые заболевания. Но возможно устранять симптомы заболевания. Цель – облегчить боль и минимизировать возможные осложнения. Также существуют лекарства, повышающие уровень гемоглобина F, что облегчает некоторые симптомы.
Однако исследования и поиск более безопасных и эффективных методов лечения все еще продолжается. В будущем для восстановления мутированного гена можно будет использовать трансплантацию стволовых клеток или генную терапию. Для того чтобы эти методы могли широко использоваться в будущем, необходимы дальнейшие обширные исследования.
Источники: БЕРТИС, Калифорния, ЭШВУД, Эр., Брунс, Делавэр, (ред.), Учебник Тиц по клинической химии и молекулярной диагностике. 4-е издание Луи: Эльзевье-Сондерс, 2006; LOTHAR, T. Клиническая лабораторная диагностика. Франкфурт: TH-Books, 1998; MASOPUST, J. Клиническая биохимия – требования и оценка биохимических исследований, часть I. и часть 2, Прага: Каролинум, 1998; RACEK, J., et al. Клиническая биохимия. 2. переработанное издание, Прага: Гален, 2006; Каспер Д.Л., Браунвальд Э., Фаучи А.С., Хаузер С.Л., Лонго Д.Л., редакторы Джеймсон Д.Л., 2005.