Норма фенилаланина в крови при фенилкетонурии 10 лет

«Помню, когда ей было три месяца, она лежала в своей маленькой корзинке на прогулочной палубе корабля. Пока мы путешествовали, я приносила ее сюда, чтобы она дышала утренним воздухом. Люди, прогуливающиеся по палубе, останавливались взглянуть на нее, и меня одолевала гордость, когда они говорили о ее необычной красоте и о разуме в ее глубоких голубых глазах», — так писала о своей первой дочери Кэрол — американская писательница Перл Бак (“The Child Who Never Grew”, 1950). Автор длительно вынашивала идею написать это произведение не только для того, чтобы выразить свою боль, но и помочь другим родителям, находящимся в подобной ситуации. Но можно сказать, что эта новелла стала, вероятно, первым описанием ребенка с далеко не редкой болезнью: в 1960 году Кэрол, сильно отстающей в развитии и обучающейся в специальной школе, поставили диагноз «фенилкетонурия».
Хотя все началось несколько раньше…
В 1934 году физиолог Асбьерн Феллинг, изучавший метаболические расстройства, определил причину необычного запаха мочи у двух норвежских детей с умственной отсталостью: виной тому был избыточный уровень одного из метаболитов фенилаланина — фенилпировиноградной кислоты. Год спустя британцем Пенроузом был предложен термин «фенилкетонурия», а также определен аутосомно-рецессивный тип передачи заболевания. Помимо этого, Пенроуз предложил лечебную диету, но она не была принята. Аналогичная идея, озвученная Джервисом и Бикелем несколько позже, уже в 50-х, стала и остается до сих пор краеугольным камнем в лечении ФКУ. В 60-х микробиолог Роберт Гатри предложил диагностический тест для определения гиперфенилаланинемии: в качестве индикатора он использовал колонии Bacillus subtilis, которым для роста необходим фенилаланин. В наши дни многие страны по всему миру включили тест Гатри (либо более новые тестовые системы, основанные на тандемной масс-спектрометрии) в программы неонатального скрининга, что позволило сразу же приступить к лечению новорожденных и избежать серьезных нарушений интеллекта. Последние 20 лет прошлого века пролили свет на генетическую природу ФКУ, а в конце первого десятилетия 21-го века была сформирована база данных мутаций гена фермента фенилаланингидроксилазы, являющихся причиной развития заболевания. Примерно в это же время были установлены генетические причины нарушения метаболизма тетрагидробиоптерина.
Итак…
Фенилкетонурия (ФКУ) — врожденное нарушение метаболизма фенилаланина, приводящее к избыточному накоплению в биологических жидкостях фенилаланина (гиперфенилаланинемии, ГФА) и его дериватов.
Наиболее часто (~ 97–98 %) развитие ФКУ обусловлено мутацией гена фенилаланингидроксилазы (ФАГ), локализованного на длинном плече 12 хромосомы, участке 12q22–q24.1, которая наследуется аутосомно-рецессивно. Данный фермент лимитирует реакцию превращения фенилаланина в тирозин, и уровень ГФА, и, соответственно, тяжесть заболевания напрямую зависят от его активности, которая определяется особенностями мутации гена.
В остальных ~ 2–3 % случаев ФКУ вызвана недостаточностью тетрагидробиоптерина, которая развивается из-за мутацией гена одного или нескольких ферментов, регулирующих его обмен (BH4-дефицитная ФКУ). BH4 является коферментом ФАГ, а также некоторых других энзимов, опосредующих синтез дофамина и серотонина (см. рис.1).
В МКБ-10 выделяют «классическую ФКУ» и «другие гиперфенилаланинемии».
«Классический» вариант заболевания дифференцируется по степени тяжести согласно уровню фенилаланина в крови (см. табл.1)
Таблица 1 | Классификация классической ФКУ по степени тяжести
| Форма ФКУ* | Уровень фенилаланина в крови, мкмоль/л | Уровень фенилаланина в крови, мг/дл |
| Легкая ГФА** (не ФКУ) | 120–600 | 2–10 |
| Умеренная (мягкая, средняя) | 600–1200 | 10–20 |
| Классическая (тяжелая) | >1200 | >20 |
ФКУ* — фенилкетонурия; ГФА** — гиперфенилаланинемия
Благодаря результатам генетических исследований была создана классификация, отражающая этиопатогенез ГФА и ФКУ (см. табл.2)
Таблица 2 | Этиопатогенетическая классификация фенилкетонурии и гиперфенилаланинемии
| Название | Причинный фермент |
| ФАГ*-зависимая ФКУ** | Фенилаланин-4-гидроксилаза |
| ГФА***, BH4****-дефицит, тип А (ФКУ, 3 типа) | 6-пирувоил-тетрагидроптерин синтаза |
| ГФА, BH4-дефицит, тип B | Гуанозинтрифосфат-циклогидролаза |
| ГФА, BH4-дефицит, тип C (ФКУ, 2 типа) | Дигидроптеридинредуктаза |
| ГФА, BH4-дефицит, тип D | Птерин-4-альфа-карбиноламиндегидратаза |
| ГФА, BH4-дефицит | Сепиаптеринредуктаза |
ФАГ* — фенилаланингидроксилаза; ФКУ** — фенилкетонурия; ГФА*** — гиперфенилаланинемия; BH4**** — тетрагидробиоптерин
Другие ГФА встречаются как при физиологических, так и при патологических состояниях. У новорожденных может быть транзиторное повышение уровня фенилаланина в крови до патологических значений ввиду незрелости ферментных систем печени или избыточного белкового питания матери, но, как правило, состояние это не длительно, а клинические проявление незначительны либо вовсе отсутствуют. Патологическая ГФА может сопровождать поражения печени различной этиологии и в этом случае будет имеет вторичный характер.
Патогенез
Фенилаланин является незаменимой аминокислотой, поступающей в организм человека преимущественно в составе белковых продуктов животного происхождения. Большая часть этой аминокислоты расходуется на синтез собственных белков организма, а оставшаяся часть — на синтез тирозина, что является главным путем катаболизма фенилаланина. Эта реакция регулируется ферментом ФАГ при участии кофермента BH4 (см. рис 1). Отсутствие данного энзима либо его малое количество (при ФКУ от 0 до 50 % нормальной активности фермента) приводит к накоплению фенилаланина и развитию клинической картины ФКУ различной степени тяжести. Не утилизированный фенилаланин катаболизируется по минорному пути с образованием токсичных продуктов (фенилацетата, фенилпирувата,фениллактата), а сниженное образование тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов (меланинов). Помимо участия в синтезе тирозина, BH4 является коферментом в реакциях образования ДОФА и серотонина.
Также на количество медиаторов ЦНС влияет и само количество фенилаланина. Дело в том, что в норме фенилаланин, а также тирозин (как уже было обозначено выше — предшественник дофамина, норадреналина и адреналина) и триптофан (предшественник серотонина) преодолевают гематоэнцефалический барьер при помощи переносчика больших нейтральных аминокислот LAT1. Возросший при ФКУ уровень фенилаланина может ингибировать LAT1, препятствуя поступлению иных субстратов в нейроны.
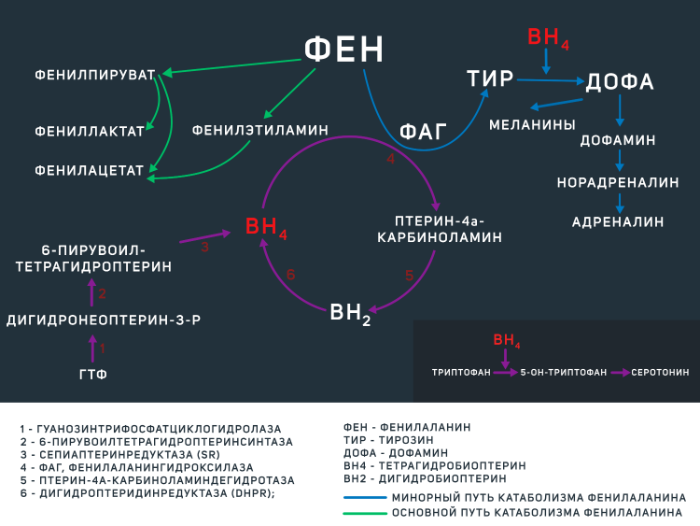
Рисунок 1 | метаболизм фенилаланина
Клиническая картина
Первые симптомы нелеченной ФАГ-зависимой ФКУ появляются, как правило, на первом году жизни ребенка, достигая максимума ко второму полугодию. Сперва обращает на себя внимание вялость ребенка либо, напротив, его беспокойство, возбужденность и срыгивания, нарушение мышечного тонуса, судороги, а также специфический затхлый запах мочи, названный «мышиным». Кроме того, нередко ФКУ проявляется эпилептическими приступами в виде абсансов, кивков, генерализованных судорог. Несколько позже, по мере роста ребенка, становится очевидным его задержка в моторном и нервно-психическом развитии. Болезнь, при отсутствии лечения, прогрессирует медленно, но неуклонно, приводя к глубокой олигофрении, несформированности речи, отсутствию игровой и предметной деятельности. Фенотипически для детей и взрослых, больных ФКУ, характерна гипопигментации кожи, волос и радужки.
При BH4-дефицитной ФКУ, помимо вышеобозначенных признаков, из-за большей недостаточности нейротрансмиттеров ЦНС выявляются атаксия, тремор, нарушения мышечного тонуса, гипокинезия, нарушения терморегуляции, затруднение глотания и поперхивания.
Диагностика
Первый этап лабораторной диагностики проводится на 3–7-й день жизни (но не ранее, чем через 2 дня от начала энтерального питания) новорожденного в рамках неонатального скрининга путем определения уровня фенилаланина на сухом пятне крови с помощью флюориметрии или тандемной масс-спектрометрии. При ГФА (фенилаланин > 120 мкмоль/л или > 2 мг/дл) проводится ретест. Если при повторном исследовании были получены подобные результаты, переходят ко второму этапу — определению отношения фенилаланин/тирозин. Этот косвенный метод позволяет провести дифференциальную диагностику между ФАГ-зависимой и BH4-зависимой ФКУ, что важно для назначения правильного лечения. Кроме лабораторных методов с целью уточнения типа заболевания используют молекулярно-генетические методы.
При отсутствие возможности провести неонатальный скрининг, в постановке диагноза опираются на клиническую картину, биохимические показатели, генеалогический анамнез, молекулярно-генетическую диагностику.
При выявлении легкой ГФА необходимо дальнейшее наблюдение и повторная диагностика.
Лечение
Основная цель терапии ФКУ — снижение уровня фенилаланина в крови для избежания нарушения моторного и нервно-психического развития ребенка — , достигается следующими методами:
- Гипофенилаланиновая диета — основной способ лечения уже более 60 лет. Для уменьшения поступления фенилаланина больным следует ограничивать прием высокобелковой пищи (мясо, рыба, яйца, молочные продукты, орехи, бобовые и др.) и вводить в рацион растительные продукты с высоким содержанием тирозина. Строгость диеты напрямую зависит от степени ГФА, меню должно составляться с опорой на факт «1 г белка = ~ 50 мг фенилаланина», возрастные физиологические нормы потребности в фенилаланине, тирозине и соотношение Б/Ж/У. У детей первого года жизни возможно употребление женского молока или молочных смесей при соответствующем расчете рациона и строгом контроле уровня фенилаланина в крови. Для восполнения недостающего белка используются аминокислотные смеси с низким содержанием фенилаланина и высоким содержанием тирозина, у детей старшего возраста компенсация происходит за счет растительных продуктов. Большой недостаток данного способа лечения — низкий комплаенс, особенно у детей подросткового возраста. Но при хорошей приверженности пациентов к диете снижение IQ можно свести к минимуму.
Некоторыми исследователями были получены данные об эффективности применения гликомакропептидов в диете. Гликомакропептиды (GLP, glycomacropeptides) — белки, получаемые из молочной сыворотки, которая богата валином, изолейцином, треонином и при этом содержит низкий уровень фенилаланина. Их использование позволило бы сделать гипофенилаланиновую диету более физиологичной, но для широкого применения необходимы дальнейшие исследования и подтверждение безопасности применения GLP в течение длительного срока. - Заместительная терапия BH4. Из-за участия BH4 в нескольких важных реакциях у больных BH4-зависимой формой ФКУ даже при хорошем соблюдении гипофенилаланиновой диеты остается симптоматика заболевания. В таком случае, как только на втором этапе лабораторной диагностики и/или на этапе медико-генетической диагностики подтверждается диагноз BH4-зависимой ФКУ, больным проводится тест на потенциальную чувствительность к сапроптерину дигидрохлориду — синтетическому аналогу BH4.
Иные методы лечения, имеющие потенциал:
- Большие нейтральные аминокислоты (The LNAAs, large neutral amino acids). Как было указано выше, фенилаланин способен конкурировать с другими аминокислотами (тирозин, триптофан) при взаимодействии с переносчиком LAT1. Некоторыми авторами было предположено, что в слизистой кишечника имеется подобный механизм, и при увеличении концентрации LNAAs всасывание фенилаланина будет уменьшаться.
- Генная терапия. Этот метод лечения мог бы стать идеальным решением, но в данный момент был тестирован лишь на мышах и требует дальнейшей серьезной разработки.
- Энзимотерапия фенилаланинамиаклиазой (PAL, phenylalanine ammonia-lyase). PAL — это фермент растений и дрожжевых грибков, осуществляющий катаболизм фенилаланина по альтернативному пути с образованием транс-циннамата и аммиака. За три последних десятилетия на мышах изучалось влияние PAL, внедренного в организм животного различными путями, начиная от оральных и инъекционных препаратов вплоть до помещения в кишечник генномодифицированных амеб, но, как и в случае с генной терапией, этот способ лечения требует дальнейшего изучения и разработки.
Источники:
- Blau N. et al. Phenylketonuria. // Lancet. Vol 376 October 23, 2010: pp 1417-1427.
- Blau N. Genetics of Phenylketonuria: Then and Now. // Human mutation, Vol 37, No. 6, 2016: pp 508-515.
- Hafid N.A., Christodoulou J. Phenylketonuria: a review of current and future treatments. // Translational Pediatrics 2015, 4(4): 304-317.
- Skirlou E., Lichter-Konecki U. Inborn Errors of Metabolism with Cognitive Impairment Metabolism Defects of Phenylalanine, Homocysteine and Methionine, Purine and Pyrimidine, and Creatine. // Pediatric Clinics of North America, Vol 65, 2018: pp 267-277.
- Руководство по педиатрии / [под ред. А.А. Баранова и др.] – Т: Врожденные и наследственные заболевания / [под ред. П.В.Новикова] – М.: “Династия”, 2007.
- Е.С. Северин и др.. Биологическая химия — М.: ООО «Медицинское информационное агентство», 2008.
- Клинические рекомендации “Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей”, 2017. https://www.pediatr-russia.ru/news/recomend
Источник
Фенилкетонурия — врожденное аутосомно-рецессивное нарушение с мутацией в хромосоме 12. Отсутствие активности фенилаланингидроксилазы в печени приводит к накоплению фенилаланина и его метаболитов в крови, моче, ликворе; при этом тирозин и катехоламины в дефиците.
Фенилкетонурия всегда сопровождается умственной отсталостью. В США 1 : 50 белых является носителем и 1:12000 страдает фенилкетонурией.
В норме содержание фенилаланина в крови 2 мг/дл.
Результаты анализов при фенилкетонурии
- Классическая форма фенилкетонурии: высокий уровень фенилаланина в крови (обычно более 30 мг/дл, в раннем детстве более 20 мг/дл), повышено содержание фенилаланина и его метаболитов в моче; уровень тирозина в норме или ниже нормы.
- Среднетяжелые формы фенилкетонурии: уровень фенилаланина в крови 15-30 мг/дл и в моче всегда присутствуют метаболиты (частота присутствия 1 : 15000).
- Легкая персистирующая гиперфенилаланинемия: уровень фенилаланина в крови 2-12 мг/дл и метаболитов в моче может не быть (частота 1 : 30000).

Для скрининга новорожденных на фенилкетонурию при уровне фенилаланина в крови менее 15 мг/дл количество фенилпируватной кислоты в моче может быть недостаточным для подтверждения диагноза колориметрическим методом. Кроме того, фенилпирувата может не быть в моче первые 2-3 недели жизни.
Ложно-отрицательный результат может быть получен в тесте Гатри со штаммами микроорганизмов, если кровь собирать в капиллярную трубку, а не на фильтровальную бумагу. Предварительные тесты позволяют определить только уровень фенилаланина более 4 мг/дл. Диагностика фенилкетонурии достовернее при нагрузочном тесте.
Если повторные тесты положительные, то проводят количественный тест уровня фенилаланина и тирозина в крови для подтверждения фенилаланинемии и исключения преходящей формы тирозинемии, которая часто встречается у новорожденных с положительными предварительными тестами. Для этого используют масс-спектрометрию и флуорометрию.
Серийные обследования должны быть проведены и у пациентов с нелеченными пограничными состояниями, потому что впоследствии в результате стресса или инфекции болезнь может проявиться.
Диагноз фенилкетонурии дополнительно подтверждают оценкой уровня метаболитов в крови и моче после нагрузочного теста: прием 100 мг аскорбиновой кислоты и забор материала спустя 24 часа.
Успешность диетотерапии при фенилкетонурии контролируется в течение жизни по уровню фенилаланина в крови:
- До 12 лет уровень фенилаланина 2-6 мг/дл.
- После 12 лет: 2-10 мг/дл.
- В юности: 2-15 мг/дл.
В течение беременности норма фенилаланина 2-5 мг/дл, потому что с повышением уровня фенилаланина в сыворотке возрастает риск микроцефалии, врожденных аномалий сердца, умственной отсталости.
Рекомендации по частоте контроля за уровнем фенилаланина в крови:
- 1-й год жизни: 1 раз в неделю.
- с 1 года до 12 лет: 2 раза в месяц.
- после 12 лет: 1 раз в месяц.
- беременным с фенилкетонурией: 2 раза в неделю.
В процессе лечения уровень фенилаланина в крови необходимо контролировать (2 раза в неделю первые 6 месяцев, 1 раз в неделю следующие 6 месяцев, 2 раза в месяц до 18 месяцев жизни, 1 раз в месяц в дальнейшем).
Диету выверяют, контролируя уровень фенилаланина в крови (например, 10 мг/дл с последовательно отрицательным тестом в моче). У беременных с нелеченной фенилкетонурией и высоким уровнем фенилаланина в крови резко возрастает частота развития у плода умственной отсталости, врожденных пороков сердца, микроцефалии.
Поиск гетерозиготных носителей и предродовая диагностика возможны с помощью анализа ДНК.

75% гетерозигот выявляется при анализом ДНК в указанные периоды, а также при пренатальной диагностике.
У 15% пациентов с фенилкетонурией выявляются врожденные пороки сердца.
Для диагностики фенилкетонурии применяется анализ с полосками хлорида железа, под действием фенилпирувата мочи они окрашиваются.
Сравнение фенилкетонурии с преходящей тирозинемией
Субстанция | Фенилкетонурия | Преходящая тирозинемия |
| Фенилаланин сыворотки | более 15 мг/дл | более 4 мг/дл (15-20 мг/дл) |
| Тирозин сыворотки | менее 5 мг/дл (выше не бывает) | более 4 мг/дл (5-20 мг/дл) |
| Гидроксифенилуксусная кислота в моче | присутствует | отсутствует |
| Моча | фенилаланин более 100 мкг/мл | широкие колебания тирозина и его метаболитов |
Источник
Распространённость заболевания
Частота данного заболевания в разных странах отличается в разы. Так, в России рождается один больной малыш на 10 тысяч новорождённых. Этот показатель в некоторых регионах Великобритании в два раза выше – 1:5000. На Африканском же континенте дети почти не болеют фенилкетонурией. Также известно, что количество девочек среди всех больных практически вдвое превышает количество мальчиков.
Среди всех наследственных болезней фенилкетонурия является почти единственной, которую полностью удаётся нейтрализовать. На сегодняшний день такого ребёнка можно вырастить абсолютно здоровым.
Исторические данные
Открытие данного заболевания связывают с именем Ивара Асбьёрна Фёллинга — норвежского врача, в 1934 году описавшего гиперфенилаланинемию, которую он ассоциировал с задержкой умственного развития. В честь открывателя патология также известна в Норвегии под названием «болезни Фёллинга».
Впервые успешное лечение было разработано и проведено в начале 50-х годов XX века группой медиков в Бирмингемском детском госпитале в Англии под руководством Хорста Биккеля. Но настоящий успех пришёл лишь после широкого применения так называемого метода Гатри, разработанного и внедрённого в 1958 — 1961 гг. (ранняя диагностика ФКУ по высокому содержанию фенилаланина в крови у новорождённых детей).
Со временем в диагностике и лечении данного заболевания стало ясно, что за него «отвечает» единственный ген — ген фенилаланингидроксилазы. Кроме того, описаны и выделены атипичные формы болезни, разработаны новые способы лечения, в ближайшей перспективе — генотерапия.
Каковы причины фенилкетонурии?
В 98% всех случаев фенилкетонурии в её основе лежит генетический дефект, а именно мутация гена XII хромосомы, который кодирует количество фермента фенилаланин-4-гидроксилазы, отвечающего за превращение аминокислоты фенилаланина в клетках печени в тирозин. В результате снижается количество фермента, что приводит к накоплению аминокислоты и продуктов её промежуточного обмена в тканях.
Вследствие побочных путей обмена фенилаланин превращается в вещества, которых в организме не должно быть в норме: фенилмолочную и фенилпировиноградную кислоты, ортофенилацетат и фенилэтиламин. Они накапливаются в крови больного и оказывают комплексное действие:
- вызывают дефицит нейромедиаторов, передающих между клетками нервной системы нервный импульс;
- нарушают процессы жирового обмена в головном мозге;
- отравляют мозг, оказывая токсическое действие.
Это вызывает необратимое снижение интеллекта. У такого ребёнка достаточно быстро развивается олигофрения.
Остальные 2% случаев заболевания связаны с другими генетическими нарушениями и зависят от концентрации иных ферментов (дигидроптеридинредуктазы и др.). Для них характерны те же самые клинические проявления, однако они не поддаются лечению диетой.
Разновидности фенилкетонурии
Выделяют две формы заболевания:
- Классическая. В данном случае фенилкетонурия является рецессивным признаком. Она встречается у одного ребёнка примерно на десять тысяч здоровых детей. Если не принимать никаких мер, то больной человек едва ли доживёт и до тридцати лет.
- Вариативная. Она не передаётся по наследству, а вызывает её генетическая мутация. Течение заболевания в этом случае более тяжёлое, а ранняя смертность прогнозируется почти со стопроцентной вероятностью.
В зависимости от дефекта гена, который блокирует определённый фермент, принято выделять три типа патологии:
- Фенилкетонурия I типа (классическая). Вызывается генетической мутацией, которая нарушает выработку в печени фермента фенилаланингидроксилазы и превращение фенилаланина в тирозин. В 98% всех случаев заболевания диагностируется именно она.
- Фенилкетонурия II типа характеризуется генетическим дефектом, вызывающим дефицит фермента дигидробиоптеринредуктазы. В результате нарушается активность органического соединения, которое необходимо для преобразования фенилаланина. Кроме того, наблюдается также пониженное содержание витамина В9 в сыворотке крови и в спинномозговой жидкости, а он необходим для утилизации аминокислот. У больных отмечаются судороги и умственная отсталость. Смертность от данного типа болезни может наступать в двух- трёхлетнем возрасте.
- Фенилкетонурия III типа провоцируется дефицитом катализатора, нужного для синтеза тетрагидробиоптерина, который необходим для превращения фенилаланина в тирозин. Вследствие этого происходит уменьшение объёмов головного мозга, что приводит к умственной отсталости.
Существует также так называемая примаптеринурия – это атипичное течение болезни, которое возникает при лёгкой форме гиперфенилаланинемии. В настоящее время ферментный дефект этого вида фенилкетонурии не выяснен. Однако данная форма патологии характеризуется высоким количеством в моче примаптерина и его производных, при этом содержание в спинномозговой жидкости нейромедиаторных метаболитов не отклоняется от нормы.
Ещё выделяют материнскую фенилкетонурию, которая наблюдается у потомства женщин с данной патологией, не соблюдающих специальную диету. Механизм развития этой формы заболевания не изучен до конца, но известно, что без регулярного контроля уровня фенилаланина у новорождённых детей выявляется ряд патологических изменений:
- недостаточный вес головного мозга;
- недоразвитие белого вещества;
- вентрикуломегалия (увеличение размеров желудочков мозга).
Фенилкетонурия у детей этого типа провоцирует хроническую интоксикацию у плода и приводит к умственной отсталости.
Клинические признаки фенилкетонурии
Ребёнок с фенилкетонурией внешне рождается совершенно здоровым, то есть не отличается ничем от других детей. В дальнейшем, с поступлением в организм пищи, начинается попадание белка, а, значит, и аминокислоты фенилаланина. Последняя постепенно накапливается, и, как правило, уже примерно к двум месяцам жизни малыша появляются первые симптомы заболевания: беспокойство или вялость, срыгивания, изменения мышечного тонуса, отсутствие интереса к окружающему миру. Иногда срыгивания настолько обильные и частые, что может возникать подозрение на какую-либо патологию желудочно-кишечного тракта. Ребёнок из-за этого плохо набирает в весе.
Приблизительно к четырём-шести месяцам становится заметной задержка психического развития. Малыш не узнаёт родителей, не реагирует на звук, не следит за игрушкой. Чем дольше длится поступление в организм фенилаланина с едой, тем выраженнее нарушения. Резко задерживается развитие речи. Словарный запас иногда может ограничиваться лишь несколькими словами. Если не будет начато своевременное лечение, то умственные нарушения к трём-четырём годам достигнут идиотии (наиболее тяжёлая степень умственной отсталости).
Особенностью фенилкетонурии является необратимость интеллектуальных и психических изменений. Таким образом, помочь таким деткам при позднем выявлении уже нельзя – они на всю жизнь остаются умственно отсталыми.
Также заметно отстаёт и физическое развитие: такие дети позже начинают переворачиваться, держать голову, сидеть. При становлении ходьбы они широко расставляют ножки, одновременно сгибая их в тазобедренных и коленных суставах. Походка у них покачивающаяся, мелкими шажками. Дети принимают так называемую «позу портного» в положении сидя — сгибают все конечности, поджимая ноги под себя. Объём головы обычно меньше нормы. Может наблюдаться выраженная микроцефалия.
Из иных неврологических симптомов возможны изменения мышечного тонуса, а также судороги. Эпилептические приступы, как правило, появляются примерно в возрасте полутора лет и приводят к прогрессированию умственных нарушений. У некоторых больных отмечаются непроизвольные движения в конечностях. В их движениях нет согласованности и плавности, нарушается равновесие.
Помимо ряда интеллектуальных и психических изменений для фенилкетонурии характерны следующие проявления:
- специфический «мышиный» запах от ребёнка (он характерен только для фенилкетонурии и появляется из-за выделения через кожу и с мочой продуктов метаболизма фенилаланина: фенилуксусной, фенилмолочной, фенилпировиноградной кислот);
- кожные проявления: шелушение, экзема, дерматиты (возникают по той же причине);
- позднее прорезывание зубов (первые зубы у таких детей могут появиться после полутора лет, эмаль при этом недоразвита);
- нарушение пигментации: обычно у таких детей голубые глаза, светлая кожа и волосы из-за снижения количества меланина;
- вегетативные симптомы: повышенная потливость, пониженное артериальное давление, запоры;
- нередко выявляются врождённые пороки сердца.
У взрослых возможно появление дрожания в конечностях, нарушений координации, судорожных припадков, ухудшения внимания и памяти, возникновение депрессии. Подобные симптомы обычно возникают при несоблюдении специализированной диеты.
У детей с фенилкетонурией бледная кожа, очень светлые волосы и голубые глаза.
Диагностика фенилкетонурии
В случае если имеется подозрение, что кто-то из родителей является носителем гена фенилкетонурии, то это можно определить при помощи генетической экспертизы в медико-генетических центрах.
Дефект ещё на этапе беременности может быть обнаружен в результате инвазивных методов пренатальной диагностики (биопсия хориона, амниоцентез, кордоцентез).
Неонатальный скрининг
После рождения всем новорождённым проводится так называемый неонатальный скрининг. Данная процедура является эффективным методом выявления самых распространённых наследственных заболеваний: адреногенитальный синдром, врождённый гипотиреоз, галактоземия, муковисцидоз и фенилкетонурия.
У каждого ребёнка в роддоме берут несколько капель крови из пяточки. Взятие образца проводят через три часа после кормления на четвёртый день жизни у доношенных детей и на седьмой день — у недоношенных. Кровь наносится на специальный тест-бланк, который затем отправляется в лабораторию, где и проводится генетическое исследование.
В случае если в данном анализе выявляют дефектный ген, то родителей с малышом приглашают для обследования в медико-генетический центр. С целью опровержения или подтверждения диагноза используются дополнительные исследования:
- в сыворотке крови;
- в сухом пятне крови;
- копрограмма;
- потовый тест;
- ДНК-диагностика.
Для подтверждения фенилкетонурии определяется концентрация в крови фенилаланина и тирозина, активность печёночных ферментов (фенилаланингидроксилазы), проводится биохимическое исследование мочи с целью обнаружения кетоновых кислот, метаболитов катехоламинов и др.
Дополнительно используются электроэнцефалография (ЭЭГ) и магнитно-резонансная томография (МРТ) головного мозга, а также осмотр ребёнка детским неврологом.
Лечение фенилкетонурии
В нашей стране на сегодняшний день единственным эффективным способом лечения является диетотерапия. Однако атипичные формы фенилкетонурии требуют постоянного приёма тетерагидробиоптерина или же его заменителей.
Питание
Чтобы нервные клетки малыша не подвергались токсическому действию фенилаланина и его производных, необходимо из рациона полностью исключить животные белки. Если сделать это на первых неделях жизни, то мозг ребёнка останется абсолютно здоровым. Если же ограничивать белок начинать в более позднем возрасте, то удаётся несколько приостановить задержку развития, однако устранить изменения в нервных клетках и вернуть здоровье нервной системе уже не удастся.
Соблюдать диету необходимо до 16 — 18 лет. Это является обязательным условием. Также желательно и в дальнейшем контролировать количество потребляемых животных белков.
Нужно придерживаться строгих правил касаемо грудного вскармливания. Необходимо использовать для кормления только сцеженное грудное молоко и лишь в количестве, разрешённом для определённого возраста. Для этого есть специальные таблицы, в которых приведены нормы потребления фенилаланина, а также существуют формулы расчёта количества молока в день. Кормящей матери при этом нужно также придерживаться специальной диеты.
Докармливают малыша специальными смесями, которые в своём составе не содержат фенилаланина. Введение прикорма у таких детей начинают с ягодных и фруктовых соков. В качестве твёрдой пищи ребёнку предлагают овощные пюре. Используют безбелковые крупы, а также безмолочные каши на основе кукурузной или рисовой муки.
Из меню детей дошкольного и школьного возраста исключают белковые продукты. Разрешены фрукты, овощи, растительные масла, изделия из крахмала.
При составлении рациона нужно строго соблюдать возрастные нормы фенилаланина:
| Возраст | Суточное количество фенилаланина (мг/кг) |
| менее 2 мес. | 60 |
| 2 — 3 мес. | 60 — 55 |
| 3 — 6 мес. | 55 — 45 |
| 6 — 12 мес. | 45 — 35 |
| 1 — 1,5 года | 35 — 30 |
| 1,5 — 3 года | 30 — 25 |
| 3 — 6 лет | 25 — 15 |
| старше 6 лет | 15 — 10 |
Также назначают витаминно-минеральные комплексы. Необходимо, чтобы ребёнок в нормальном количестве получал витамины B1, В6, С, фолиевую кислоту, кальций, железо и магний. Количество потребляемых калорий должно быть на 30% увеличено по сравнению с возрастной нормой.
Выделяют три группы натуральных продуктов, в основе которых лежит количество содержащегося в них фенилаланина.
- Красный список – это продукты, которые полностью необходимо исключить из рациона: все виды мяса и рыбы, морепродукты, колбасные изделия, яйца, творог, сыры, хлеб и хлебобулочные изделия, орехи, крупы и хлопья, кондитерские изделия, продукты из сои.
- Оранжевый список – продукты, которые под строгим контролем разрешены в небольших количествах: молочные продукты, овощи (картофель, капуста), овощные консервы, рис и кукуруза.
- Зелёный список – продукты, которые могут употребляться без каких-либо ограничений: овощи, фрукты, зелень, ягоды, крахмал, сахар, мёд, растительное и сливочное масло, топлёный жир.
Ещё две группы продуктов выпускает промышленность:
- готовые пюре на основе фруктов для детского питания;
- низкобелковые искусственные продукты для диетического питания (печенье, макароны, хлеб).
Родителям важно уметь правильно составлять диету и рассчитывать необходимое количество фенилаланина. Для этого нужно под рукой иметь весы, которые позволяют взвешивать до десятой доли грамма.
Прогноз
Проведение неонатального скрининга позволяет предотвратить серьёзные нарушения функции печени и повреждения нервной системы. При раннем назначении специальной диеты прогноз развития детей достаточно хороший. Однако стоит помнить, что при поздно начатом лечении в отношении умственного и психического развития ребёнка прогноз неблагоприятный.
Заключение
С целью оценки вероятности рождения малыша с фенилкетонурией должны пройти предварительное генетическое консультирование супружеские пары, уже имеющие такого ребёнка в семье, состоящие в родственном браке или имеющие родственников с фенилкетонурией. А женщины с этим заболеванием обязаны соблюдать строгую диету до и во время беременности.
По окончании университета прошла интернатуру по направлению «Неонатология» в СамГМУ. После завершения профессиональной подготовки, и по настоящее время, работаю врачом-неонатологом в ГУЗ Городская клиническая больница №1 (Перинатальный центр) г. Ульяновск.
Оценка статьи
Мы приложили много усилий, чтобы Вы смогли прочитать эту статью, и будем рады Вашему отзыву в виде оценки. Автору будет приятно видеть, что Вам был интересен этот материал. Спасибо!
Загрузка…
Источник