Норма фенилаланина в крови у детей

Что такое Фенилкетонурия (ФКУ) у детей –
Фенилкетонурия (ФКУ) у детей — генетическая болезнь, которая характеризуется нарушениями обмена фенилаланина и бывает у 1 из 8000–15 000 новорожденных. Форм фенилкетонурии (ФКУ) всего 4, но существует 400 разных мутаций и метаболические фенотипы заболевания.
Фенилкетонурия — наследственная аминоацидопатия, при которой снижается интеллект ребенка, и возникает неврологический дефицит.
Фенилкетонурия I (классическая или тяжелая) – это аутосомно-рецессивное заболевание, которое возникает вследствие мутации гена фенилаланингидроксилазы. В основе заболевания лежит нехватка фенилаланин-4-гидроксилазы которая обеспечивает превращение фенилаланина в тирозин, результатом чего становится накопление фенилаланина и его метаболитов в тканях и физиологических жидкостях организма ребенка.
Отдельную группу представляю атипичные варианты фенилкетонурии. При них симптомы очень похожи на таковые при классическом варианте заболевания. Но нет положительных продвижений по показателям развития ребенка, даже если проводить нужную диетотерапию. Такие варианты объясняются нехваткой дегидроптеринредуктазы, тетрагидроптерина, гуанозин-5-трифосфатциклогидролазы, 6-пирувоилтетрагидроптеринсинтазы и пр.
Фенилкетонурия II (атипичная) — аутосомно-рецессивная болезнь, при которой генный дефект находится в коротком плече хромосомы 4. Характеризуется она нехваткой дегидроптеринредуктазы, что приводит к нарушению восстановления активной формы тетрагидробиоптерина, а в спинномозговой жидкости и сыворотке крови снижается уровень фолатов. Результат таких изменений – метаболические блоки в механизмах превращения фенилаланина в тирозин. Заболевание было выявлено еще в конце 20-го века.
Фенилкетонурия III (атипичная) — аутосомно-рецессивная болезнь, которая вызвана недостаточностью 6-пирувоилтетрагидроптеринсинтазы. Он принимает участие в организме в процессе создания тетрагидробиоптерина из дигидронеоптеринтрифосфата, что было открыто в конце 20-го столетия. Нарушения сходы с таковыми при выше описанной (второй) форме.
Примаптеринурия — атипичная фенилкетонурия у детей с легкой гиперфенилаланинемией, у которых присутствует в больших количества в моче примаптерин и часть его производных, а в спинномозговой жидкости нормальная концентрация нейромедиаторных метаболитов.
Материнская ФКУ – болезнь, при которой снижается уровень интеллекта (вплоть до умственной отсталости) среди потомства женщин, которые больны фенилкетонурией и не сидели на специальной идете, когда были совершеннолетними.
Есть предположения, что при материнской ФКУ нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита. Было проведено исследование в 2008 году Кочем и его командой. У младенца, рожденного от матери с ФКУ, при аутопсии головного было найдено некоторое количество патологических изменений: вентикуломегалия, низкий вес мозга, задержка миелинизации (признаков астроцитоза не наблюдалось), гипоплазия белого вещества.
В некоторых странах СНГ применяется условная классификация рассматриваемого заболевания по уровню содержания в сыворотке крови фенилаланина:
Название формы | Уровень фенилаланина |
классическая | выше 20 мг% (1200 мкмоль/л) |
средняя | 10,1–20 мг% (600–1200 мкмоль/л) |
легкая | до 8 мг% (480 мкмоль/л) |
 у детей")
Что провоцирует / Причины Фенилкетонурии (ФКУ) у детей:
Основная причина фенилкетонурии у детей – нехватка фермента с названием фенилаланин-4-гидроксилаз. Из-за его отсутствия происходит скопление в жидкостях и тканях фенилаланина в большом количестве, который воздействует токсически на ЦНС ребенка. Результатом становится нарушения метаболизма гормонов, обмена белков, транспорта аминокислот, обмена глико- и липопротеидов.
Причиной ФКУ у детей может стать:
- хронический алкоголизм матери и/или отца
- инфекционные воспалительные процессы половых органов родителей
- влияние на организм отца и матери пагубных факторов и пр.
Патогенез (что происходит?) во время Фенилкетонурии (ФКУ) у детей:
Основа патогенеза фенилкетонурии (ФКУ) у детей является нехватка фермента фенилаланин-4-гидроксилаза, который обеспечивает превращение фенилаланина в тирозин. Производные фенилаланина (фенилпировиноградная, фенилуксусная, фенилмолочная кислоты, фенилэтилламин, фенилацетилглютамин) и само вещество скапливаются в тканях. Следствием является негативное влияние на центральную нервную систему. Возникают разные нарушения, в том числе в обмене катехоламинов и серотонина.
Симптомы Фенилкетонурии (ФКУ) у детей:
Новорожденный ребенок не похож на больного. Симптомы фенилкетонурии (ФКУ) начинают быть заметны в возрасте 2-6 месяцев. Типичные проявления:
- отсутствие интереса к окружающему миру
- выраженная вялость
- рвота
- беспокойство
- повышенная раздражительность
С 6 месяцев у малыше заметно отставание в психическом развитии. У меньшинства детей это олигофрения в слабой степени. А более чем у половины детей фиксируют идитию. Рост малыша с ФКУ может быть нормальным или сниженным. Зубки режутся поздно, череп может иметь размеры меньше нормы. Сидеть и ходить ребенок с фенилкетонурией начинает поздно.
Детей с рассматриваемым диагнозом можно отличить по позе и походке. Они широко расставляют ноги, сгибая их в тазобедренном и коленных суставах. Шаги мелкие. При ходьбе ребенок покачивается. Сидят они в так называемом положении портного – поджав ноги, поскольку у них повышен мышечный тонус.
При фенилкетонурии (ФКУ) дети обычно имеют голубой цвет глаз и светлый оттенок волос. Кожа почти не пигментирована. От ребенка слышен «мышиный» запах. В некоторых случаях у больного могут быть припадки эпилепсии, но они проходят по мере взросления ребенка.
Другие типичные симптомы ФКУ у детей:
- дермографизм
- потливость
- повышенная чувствительность к солнечным лучам и травмам
- акроцианоз
- тяжёлая экзема
- дерматит
- склонность к запорам
- артериальная гипотония
- расстройства аутистического спектра
- гиперактивность
Если не провести вовремя лечение, уровень интеллекта ребенка будет составлять менее 50. В возрасте 18 месяцев могут появиться судорожные приступы. Исчезают они спонтанно. В раннем возрасте приступы часто проходят в форме инфантильных спазмов, далее становятся тоникоклоническими припадками.
Диагностика Фенилкетонурии (ФКУ) у детей:
Для диагностики фенилкетонурии (ФКУ) у детей определяют содержание крови уровней фенилаланина и тирозина в крови. Применяют тест Гатри, пробу Феллинга, флуориметрию, хроматографию, МРТ, поиск мутантного гена, электроэнцефалографию.
ЭЭГ позволяет обнаружить нарушения в основном в виде паттерна гипсартимии, даже если приступов у ребенка не наблюдалось. Также находят фокусы спайк- и полиспайк-разрядов (единичные и множественные). МРТ не находит изменений сигнала в стволе, мозжечке или коре головного мозга. Изменения на МРТ не коррелируют с уровнем интеллекта, они зависят от содержания фенилаланина в крови.
Если у ребенка фенилкетонурия II, то симптомы проявляются после 12 месяцев жизни. В крови затем находят повышенный уровень фенилаланина в периоде новорожденности, назначают диету, но болезнь всё равно прогрессирует. У малышей выраженная умственная отсталость, судороги, признаки повышенной возбудимости, гиперрефлексия, мышечная дистония, спастический тетрапарез. Летальный исход в части случаев наступает в возрасте от 2 до 3 лет.
Симптомы фенилкетонурии III напоминают выше перечисленные. У ребенка врачи обнаруживают три типичных признака:
- спастический тетрапарез
- микроцефалия
- глубокая умственная отсталость
Лечение Фенилкетонурии (ФКУ) у детей:
Новые методы лечения
На сегодня исследователи разрабатывают несколько методов альтернативной терапии фенилкетонурии (ФКУ):
- энзимотерапия фенилаланингидроксилазой, фенилаланинаммониалиазой
- метод «больших нейтральных аминокислот»
- лечение тетрагидробиоптерином (Сапроптерин)
Существует информация о случаях, когда пациентам с умеренной или легкой формой заболевания помогал тетрагидробиоптерин в дозе от 10 до 20 мг на 1 кг тела в сутки. В 2008 году было доказано, что для нормального физического развития детей с фенилкетонурией можно применять пищевые гликомакропептиды, которые также снижают содержание в плазме крови и головном мозге фенилаланина. Экспериментальным методом считается введение непосредственно в пораженные клетки печени ребенка введение гена фенилаланингидроксилазы. Этот метод пока не актуален в странах СНГ, в том числе Украине и России.
Продукты для детей с ФКУ (фенилкетонурией)
Диетотерапия позволяет предотвратить интеллектуальный дефицит при классической форме рассматриваемой болезни. Важное значение имеет возраст малыша, когда начинают применять диету. Каждый месяц без применения диеты малыш с ФКУ теряет примерно 4 балла IQ. Вопрос о диете при фенилкетонурии у детей в различных странах рассматривают по-разному. Но принципы едины.
При уровне уровень фенилаланина в крови до 2–6 мг% (120–360 мкмоль/л) у младенцев диета не применяется. Суть питания детей с ФКУ – в продуктах с низким содержанием фенилаланина, в основном это небелковая пища. Она нужна детям первого года жизни. В более позднем возрасте такое питание приносит меньше результатов.
Лечебный рацион питания при ФКУ:
- натуральные продукты питания
- лечебные продукты
- малобелковые продукты на основе крахмала
Детям с фенилкетонурией нельзя:
- птицу
- мясо
- молочные продукты
- рыбу
- грудное молоко (детям до 12 месяцев)
Смеси детям с ФКУ нужны только с минимальным содержанием белка. В течение первого года жизни допустимое количество фенилаланина составляет от 90 до 35 мг/кг ребенка. 50 мг фенилаланина = 1 г белка.
Лечебные продукты при фенилкетонурии у детей:
- ХР Аналог LCP
- MD мил ФКУ-0
- Афенилак
Диеты ребенку нужно придерживаться, если показатель фенилаланина в крови составляет минимум 360–480 ммоль/л.
Прикорм при фенилкетонурии (ФКУ у детей)
По достижению ребенком 3-месячного возраста рацион нужно расширять, вводя фруктовые и ягодные соки. Сначала это 3-5 капель, потом объем увеличивают до 30–50 мл. Для детей 12 месяцев доза уже составляет до 100 мл.
Соки в качестве прикорма:
- грушевый
- яблочный
- сливовый
Также в рацион постепенно вводят фруктовое пюре, постепенно увеличивая порцию. Детям от 4-4,5 месяцев уже можно овощное пюре, которое готовится родителями. Также можно плодовоовощные консервы для грудничков, но без молока. Второй прикорм – каша (10%) из безбелковой крупки или саго. Также ребенку можно давать безмолочные каши промышленного производства из кукурузной и/или рисовой муки. В них должно быть меньше 1 грамма белка на 100 мл готового продукта.
Детям от 6 месяцев можно вводить в диету кисели и/или муссы, в которых нет белка. Их готовят на амилопектиновом набухающем крахмале и фруктовом соке; это низкобелковый молочный напиток PKU «Лопрофин» и безбелковый напиток с молочным вкусом Нутриген.Детям с ФКУ от 7 месяцев можно давать низкобелковые изделия «Лопрофин»: рис, спагетти, спиральки. С 8 месяцев при фенилкетонурии малышам можно давать специальный безбелковый хлеб.
Диета для ФКУ у детей от 1 года
Для питания таких пациентов применяют продукты на основе смесей аминокислот без содержания фенилаланина и/или гидролизатов белка или с мизерным его количеством. В их составле должны быть комплексы макро-, микроэлементов и витаминов. По мере взросления ребенка дозу белка можно увеличивать, но не сразу. Количество углеводов и жиров нужно постепенно снижать, а потом и вовсе исключить. Рацион постепенно расширяется за счет натуральных продуктов и блюда.
Для детей с ФКУ от 12 месяцев можно применять специализированные лечебные продукты:
- Тетрафен 40
- Тетрафен 30
- MD мил ФКУ-1
- Тетрафен 70
- MD мил ФКУ-3
- Изифен
- П-АМ 1, П-АМ 2, П-АМ 3
- ХР Максамум (вкус нейтральный или апельсиновый)
- ХР Максамейд
Врачи советуют постепенно переходить на продукты для детей более старшего возраста на протяжении 1-2 недель. Объем предыдущей смеси нужно уменьшить на 1/4–1/5 и добавить эквивалентное по белку количество нового продукта. В части лечебных продуктов содержатся полиненасыщенные жирные кислоты. Среди малобелковых продуктов зарубежного производства есть напитки без белка, десерты, соусы и приправы, печенье и специальные сорта хлеба, которые можно детям при фенилкетонурии (ФКУ).
Часть исследователей склоняется к мнению, что детям с ФКУ диету нужно обогащать тирозином. Лечебные продукты имеют специфический вкус, потому могут быть нужны вкусовые добавки без содержания белка. Нельзя применять подсластитель аспартам, потому что при расщеплении он образует в том числе фенилаланин.
При лечении нужен регулярный контроль содержания фенилаланина в крови. Детей до 3 месяцев проверяют 1 раз в неделю, а после получения стабильных результатов – минимум 1 раз в 2 недели. Детей с ФКУ от 3 мес. до 1 года проверяют 1 раз в месяц, иногда – 2 раза. Для детей от 1 до 3 лет осмотры нужны минимум 1 раз в два месяца, а после трех лет контроль проводят 1 раз в 3 месяца.
Необходим контроль таких показателей для детей с ФКУ:
- физическое и интеллектуальное развитие ребенка
- нутритивный статус больного
- эмоциональное развитие
- речевое развитие
Один раз в месяц нужно проводить общий анализ крови. А по показаниям – биохимический анализ крови.
Если у ребенка обнаруживают дополнительные болезни с диспепсическими явлениями, интоксикацией, гипертермией, то диету можно прекратить на 2-3 дня, заменив лечебные продукты на натуральные, в которых не слишком много белка. Когда острая фаза болезни заканчивается, в рацион снова вводят лечебные продукты, но быстрее, чем в начале ввода диеты.
Прекращение диетотерапии
Возраст прекращения специальной диеты при ФКУ у детей до сих пор в процессе дискуссии. Существует информация, что при отмене диетотерапии в 5-летнем возрасте у одной трети детей с ФКУ отмечалось снижение уровня IQ на 10 баллов и более на протяжении следующих 5 лет. Прекращения диеты для детей старше 15 лет в некоторых случаях сказывались на прогрессирующих изменениях белого вещества мозга, что было выявлено при помощи МРТ.
При классической фенилкетонурии у детей диеты нужно придерживаться всю жизнь. Общее количество белка после наступления совершеннолетия не должно быть больше, чем 0,8–1,0 г/кг в сутки.
Профилактика Фенилкетонурии (ФКУ) у детей:
Чтобы организовать раннюю диетотерапию и избежать тяжелых церебральных повреждений, нужно проводить массовые скрининги на фенилкетонурию в неонатальном периоде. Это позволяет также избежать нарушения функционирования печени ребенка. Чтобы оценить риск рождения ребенка с рассматриваемым диагнозом, нужно предварительное генетическое консультирование для пар, у которых уже есть ребенок с фенилкетонурией (ФКУ) или у которых есть родственники с такой болезнью.
Женщины с фенилкетонурией до момента зачатия должны строго придерживаться диеты и продолжать ее, пока будут беременными. Это позволит избежать нарушений развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Дети с ФКУ должны наблюдаться участковым педиатром и психоневрологом.
К каким докторам следует обращаться если у Вас Фенилкетонурия (ФКУ) у детей:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Фенилкетонурии (ФКУ) у детей, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу.
Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас ? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно. Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые симптомы болезни. Определение симптомов – первый шаг в диагностике заболеваний в целом. Для этого просто необходимо по несколько раз в год проходить обследование у врача, чтобы не только предотвратить страшную болезнь, но и поддерживать здоровый дух в теле и организме в целом.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации, возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой. Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе Вся медицина. Также зарегистрируйтесь на медицинском портале Eurolab, чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
Другие заболевания из группы Болезни ребенка (педиатрия):
Если Вас интересуют еще какие-нибудь виды болезней и группы заболеваний человека или у Вас есть какие-либо другие вопросы и предложения – напишите нам, мы обязательно постараемся Вам помочь.
«Помню, когда ей было три месяца, она лежала в своей маленькой корзинке на прогулочной палубе корабля. Пока мы путешествовали, я приносила ее сюда, чтобы она дышала утренним воздухом. Люди, прогуливающиеся по палубе, останавливались взглянуть на нее, и меня одолевала гордость, когда они говорили о ее необычной красоте и о разуме в ее глубоких голубых глазах», — так писала о своей первой дочери Кэрол — американская писательница Перл Бак (“The Child Who Never Grew”, 1950). Автор длительно вынашивала идею написать это произведение не только для того, чтобы выразить свою боль, но и помочь другим родителям, находящимся в подобной ситуации. Но можно сказать, что эта новелла стала, вероятно, первым описанием ребенка с далеко не редкой болезнью: в 1960 году Кэрол, сильно отстающей в развитии и обучающейся в специальной школе, поставили диагноз «фенилкетонурия».
Хотя все началось несколько раньше…
В 1934 году физиолог Асбьерн Феллинг, изучавший метаболические расстройства, определил причину необычного запаха мочи у двух норвежских детей с умственной отсталостью: виной тому был избыточный уровень одного из метаболитов фенилаланина — фенилпировиноградной кислоты. Год спустя британцем Пенроузом был предложен термин «фенилкетонурия», а также определен аутосомно-рецессивный тип передачи заболевания. Помимо этого, Пенроуз предложил лечебную диету, но она не была принята. Аналогичная идея, озвученная Джервисом и Бикелем несколько позже, уже в 50-х, стала и остается до сих пор краеугольным камнем в лечении ФКУ. В 60-х микробиолог Роберт Гатри предложил диагностический тест для определения гиперфенилаланинемии: в качестве индикатора он использовал колонии Bacillus subtilis, которым для роста необходим фенилаланин. В наши дни многие страны по всему миру включили тест Гатри (либо более новые тестовые системы, основанные на тандемной масс-спектрометрии) в программы неонатального скрининга, что позволило сразу же приступить к лечению новорожденных и избежать серьезных нарушений интеллекта. Последние 20 лет прошлого века пролили свет на генетическую природу ФКУ, а в конце первого десятилетия 21-го века была сформирована база данных мутаций гена фермента фенилаланингидроксилазы, являющихся причиной развития заболевания. Примерно в это же время были установлены генетические причины нарушения метаболизма тетрагидробиоптерина.
Итак…
Фенилкетонурия (ФКУ) — врожденное нарушение метаболизма фенилаланина, приводящее к избыточному накоплению в биологических жидкостях фенилаланина (гиперфенилаланинемии, ГФА) и его дериватов.
Наиболее часто (~ 97–98 %) развитие ФКУ обусловлено мутацией гена фенилаланингидроксилазы (ФАГ), локализованного на длинном плече 12 хромосомы, участке 12q22–q24.1, которая наследуется аутосомно-рецессивно. Данный фермент лимитирует реакцию превращения фенилаланина в тирозин, и уровень ГФА, и, соответственно, тяжесть заболевания напрямую зависят от его активности, которая определяется особенностями мутации гена.
В остальных ~ 2–3 % случаев ФКУ вызвана недостаточностью тетрагидробиоптерина, которая развивается из-за мутацией гена одного или нескольких ферментов, регулирующих его обмен (BH4-дефицитная ФКУ). BH4 является коферментом ФАГ, а также некоторых других энзимов, опосредующих синтез дофамина и серотонина (см. рис.1).
В МКБ-10 выделяют «классическую ФКУ» и «другие гиперфенилаланинемии».
«Классический» вариант заболевания дифференцируется по степени тяжести согласно уровню фенилаланина в крови (см. табл.1)
Таблица 1 | Классификация классической ФКУ по степени тяжести
| Форма ФКУ* | Уровень фенилаланина в крови, мкмоль/л | Уровень фенилаланина в крови, мг/дл |
| Легкая ГФА** (не ФКУ) | 120–600 | 2–10 |
| Умеренная (мягкая, средняя) | 600–1200 | 10–20 |
| Классическая (тяжелая) | >1200 | >20 |
ФКУ* — фенилкетонурия; ГФА** — гиперфенилаланинемия
Благодаря результатам генетических исследований была создана классификация, отражающая этиопатогенез ГФА и ФКУ (см. табл.2)
Таблица 2 | Этиопатогенетическая классификация фенилкетонурии и гиперфенилаланинемии
| Название | Причинный фермент |
| ФАГ*-зависимая ФКУ** | Фенилаланин-4-гидроксилаза |
| ГФА***, BH4****-дефицит, тип А (ФКУ, 3 типа) | 6-пирувоил-тетрагидроптерин синтаза |
| ГФА, BH4-дефицит, тип B | Гуанозинтрифосфат-циклогидролаза |
| ГФА, BH4-дефицит, тип C (ФКУ, 2 типа) | Дигидроптеридинредуктаза |
| ГФА, BH4-дефицит, тип D | Птерин-4-альфа-карбиноламиндегидратаза |
| ГФА, BH4-дефицит | Сепиаптеринредуктаза |
ФАГ* — фенилаланингидроксилаза; ФКУ** — фенилкетонурия; ГФА*** — гиперфенилаланинемия; BH4**** — тетрагидробиоптерин
Другие ГФА встречаются как при физиологических, так и при патологических состояниях. У новорожденных может быть транзиторное повышение уровня фенилаланина в крови до патологических значений ввиду незрелости ферментных систем печени или избыточного белкового питания матери, но, как правило, состояние это не длительно, а клинические проявление незначительны либо вовсе отсутствуют. Патологическая ГФА может сопровождать поражения печени различной этиологии и в этом случае будет имеет вторичный характер.
Патогенез
Фенилаланин является незаменимой аминокислотой, поступающей в организм человека преимущественно в составе белковых продуктов животного происхождения. Большая часть этой аминокислоты расходуется на синтез собственных белков организма, а оставшаяся часть — на синтез тирозина, что является главным путем катаболизма фенилаланина. Эта реакция регулируется ферментом ФАГ при участии кофермента BH4 (см. рис 1). Отсутствие данного энзима либо его малое количество (при ФКУ от 0 до 50 % нормальной активности фермента) приводит к накоплению фенилаланина и развитию клинической картины ФКУ различной степени тяжести. Не утилизированный фенилаланин катаболизируется по минорному пути с образованием токсичных продуктов (фенилацетата, фенилпирувата,фениллактата), а сниженное образование тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов (меланинов). Помимо участия в синтезе тирозина, BH4 является коферментом в реакциях образования ДОФА и серотонина.
Также на количество медиаторов ЦНС влияет и само количество фенилаланина. Дело в том, что в норме фенилаланин, а также тирозин (как уже было обозначено выше — предшественник дофамина, норадреналина и адреналина) и триптофан (предшественник серотонина) преодолевают гематоэнцефалический барьер при помощи переносчика больших нейтральных аминокислот LAT1. Возросший при ФКУ уровень фенилаланина может ингибировать LAT1, препятствуя поступлению иных субстратов в нейроны.
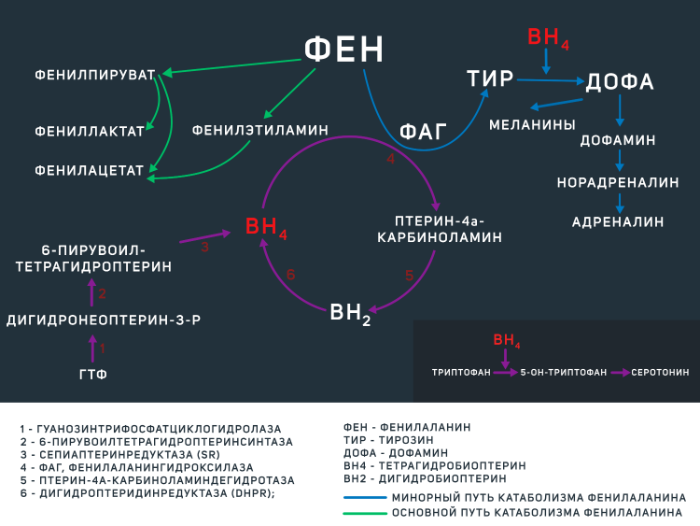
Рисунок 1 | метаболизм фенилаланина
Клиническая картина
Первые симптомы нелеченной ФАГ-зависимой ФКУ появляются, как правило, на первом году жизни ребенка, достигая максимума ко второму полугодию. Сперва обращает на себя внимание вялость ребенка либо, напротив, его беспокойство, возбужденность и срыгивания, нарушение мышечного тонуса, судороги, а также специфический затхлый запах мочи, названный «мышиным». Кроме того, нередко ФКУ проявляется эпилептическими приступами в виде абсансов, кивков, генерализованных судорог. Несколько позже, по мере роста ребенка, становится очевидным его задержка в моторном и нервно-психическом развитии. Болезнь, при отсутствии лечения, прогрессирует медленно, но неуклонно, приводя к глубокой олигофрении, несформированности речи, отсутствию игровой и предметной деятельности. Фенотипически для детей и взрослых, больных ФКУ, характерна гипопигментации кожи, волос и радужки.
При BH4-дефицитной ФКУ, помимо вышеобозначенных признаков, из-за большей недостаточности нейротрансмиттеров ЦНС выявляются атаксия, тремор, нарушения мышечного тонуса, гипокинезия, нарушения терморегуляции, затруднение глотания и поперхивания.
Диагностика
Первый этап лабораторной диагностики проводится на 3–7-й день жизни (но не ранее, чем через 2 дня от начала энтерального питания) новорожденного в рамках неонатального скрининга путем определения уровня фенилаланина на сухом пятне крови с помощью флюориметрии или тандемной масс-спектрометрии. При ГФА (фенилаланин > 120 мкмоль/л или > 2 мг/дл) проводится ретест. Если при повторном исследовании были получены подобные результаты, переходят ко второму этапу — определению отношения фенилаланин/тирозин. Этот косвенный метод позволяет провести дифференциальную диагностику между ФАГ-зависимой и BH4-зависимой ФКУ, что важно для назначения правильного лечения. Кроме лабораторных методов с целью уточнения типа заболевания используют молекулярно-генетические методы.
При отсутствие возможности провести неонатальный скрининг, в постановке диагноза опираются на клиническую картину, биохимические показатели, генеалогический анамнез, молекулярно-генетическую диагностику.
При выявлении легкой ГФА необходимо дальнейшее наблюдение и повторная диагностика.
Лечение
Основная цель терапии ФКУ — снижение уровня фенилаланина в крови для избежания нарушения моторного и нервно-психического развития ребенка — , достигается следующими методами:
- Гипофенилаланиновая диета — основной способ лечения уже более 60 лет. Для уменьшения поступления фенилаланина больным следует ограничивать прием высокобелковой пищи (мясо, рыба, яйца, молочные продукты, орехи, бобовые и др.) и вводить в рацион растительные продукты с высоким содержанием тирозина. Строгость диеты напрямую зависит от степени ГФА, меню должно составляться с опорой на факт «1 г белка = ~ 50 мг фенилаланина», возрастные физиологические нормы потребности в фенилаланине, тирозине и соотношение Б/Ж/У. У детей первого года жизни возможно употребление женского молока или молочных смесей при соответствующем расчете рациона и строгом контроле уровня фенилаланина в крови. Для восполнения недостающего белка используются аминокислотные смеси с низким содержанием фенилаланина и высоким содержанием тирозина, у детей старшего возраста компенсация происходит за счет растительных продуктов. Большой недостаток данного способа лечения — низкий комплаенс, особенно у детей подросткового возраста. Но при хорошей приверженности пациентов к диете снижение IQ можно свести к минимуму.
Некоторыми исследователями были получены данные об эффективности применения гликомакропептидов в диете. Гликомакропептиды (GLP, glycomacropeptides) — белки, получаемые из молочной сыворотки, которая богата валином, изолейцином, треонином и при этом содержит низкий уровень фенилаланина. Их использование позволило бы сделать гипофенилаланиновую диету более физиологичной, но для широкого применения необходимы дальнейшие исследования и подтверждение безопасности применения GLP в течение длительного срока. - Заместительная терапия BH4. Из-за участия BH4 в нескольких важных реакциях у больных BH4-зависимой формой ФКУ даже при хорошем соблюдении гипофенилаланиновой диеты остается симптоматика заболевания. В таком случае, как только на втором этапе лабораторной диагностики и/или на этапе медико-генетической диагностики подтверждается диагноз BH4-зависимой ФКУ, больным проводится тест на потенциальную чувствительность к сапроптерину дигидрохлориду — синтетическому аналогу BH4.
Иные методы лечения, имеющие потенциал:
- Большие нейтральные аминокислоты (The LNAAs, large neutral amino acids). Как было указано выше, фенилаланин способен конкурировать с другими аминокислотами (тирозин, триптофан) при взаимодействии с переносчиком LAT1. Некоторыми авторами было предположено, что в слизистой кишечника имеется подобный механизм, и при увеличении концентрации LNAAs всасывание фенилаланина будет уменьшаться.
- Генная терапия. Этот метод лечения мог бы стать идеальным решением, но в данный момент был тестирован лишь на мышах и требует дальнейшей серьезной разработки.
- Энзимотерапия фенилаланинамиаклиазой (PAL, phenylalanine ammonia-lyase). PAL — это фермент растений и дрожжевых грибков, осуществляющий катаболизм фенилаланина по альтернативному пути с образованием транс-циннамата и аммиака. За три последних десятилетия на мышах изучалось влияние PAL, внедренного в организм животного различными путями, начиная от оральных и инъекционных препаратов вплоть до помещения в кишечник генномодифицированных амеб, но, как и в случае с генной терапией, этот способ лечения требует дальнейшего изучения и разработки.
Источники:
- Blau N. et al. Phenylketonuria. // Lancet. Vol 376 October 23, 2010: pp 1417-1427.
- Blau N. Genetics of Phenylketonuria: Then and Now. // Human mutation, Vol 37, No. 6, 2016: pp 508-515.
- Hafid N.A., Christodoulou J. Phenylketonuria: a review of current and future treatments. // Translational Pediatrics 2015, 4(4): 304-317.
- Skirlou E., Lichter-Konecki U. Inborn Errors of Metabolism with Cognitive Impairment Metabolism Defects of Phenylalanine, Homocysteine and Methionine, Purine and Pyrimidine, and Creatine. // Pediatric Clinics of North America, Vol 65, 2018: pp 267-277.
- Руководство по педиатрии / [под ред. А.А. Баранова и др.] – Т: Врожденные и наследственные заболевания / [под ред. П.В.Новикова] – М.: “Династия”, 2007.
- Е.С. Северин и др.. Биологическая химия — М.: ООО «Медицинское информационное агентство», 2008.
- Клинические рекомендации “Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей”, 2017. https://www.pediatr-russia.ru/news/recomend