Рефрактерная анемия дифференциальная диагностика

Дифференциальная диагностика миелодиспластических синдромов (МДС)Следует иметь в виду, что при миелодиспластических синдромах (МДС) не существует патогномоничных признаков, и диагноз устанавливают с учетом анамнеза, данных осмотра, но прежде всего на основании комплекса лабораторных исследований. К обязательным методам обследования при установлении диагноза миелодиспластического синдрома (МДС) относятся: Согласно классификации ВОЗ, введена новая рубрика «Миелодиспластические/миелопролиферативные заболевания», которая включает в себя: хронический миеломоноцитарный лейкоз (ХММЛ), атипичный хронический миелоидный лейкоз (АХМЛ), ювенильный миеломоноцитарный лейкоз (ЮММЛ), миелодиспластическое/миелопролиферативное заболевание неклассифицируемое (МДС/МПЗ-Н). Ввиду того, что ХММЛ ранее рассматривался в группе МДС и в связи с возможными дифференциально диагностическими трудностями, мы приводим дигностические критерии миелодиспластических/миелопролиферативных заболеваний за исключением ЮММЛ, поскольку он встречается в детском возрасте. Диагностическими критериями ХММЛ являются: 4) дисплазия клеток одного и более ростков кроветворения. При отсутствии дисплазии или ее минимальных проявлениях диагноз ХММЛ может быть установлен, если обнаружены: В связи с разным прогнозом в зависимости от числа бластных клеток ХММЛ разделен на два подварианта: ХММЛ-1 (с числом бластных клеток в крови менее 5 %, в костном мозге — менее 10%), ХММЛ-2 (с числом бластных клеток в крови от 5 до 19 %, в костном мозге — от 10 до 19 %). При наличии палочек Ауэра в бластных клетках и количестве бластов в крови и костном мозге менее 20 % устанавливается диагноз ХММЛ-2. Случаи ХММЛ с эо-зинофилией выше 1,5• 109/л классифицируют как ХММЛ-1 или ХММЛ-2 с эозинофилией.
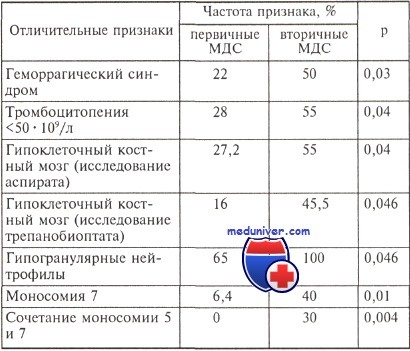
Дифференциально-диагностические сложности могут возникать между миелодиспластическим синдромом (МДС) и АХМЛ. Диагноз АХМЛ устанавливают при сочетании следующих признаков: Частота обнаружения цитогенетических аномалий при АХМЛ может достигать 80 %, однако специфичные аберрации отсутствуют. Прогноз неблагоприятный: медиана выживаемости не превышает 20 мес, а до трансформации заболевания в острый лейкоз доживают от 25 до 40 % больных. К признакам МДС/МПЗ-Н относят: Поскольку при рефрактерной анемии с кольцевыми сидеробластами может отмечаться тромбоцитоз свыше 600,0•109/л, авторы классификации ВОЗ считают, что в подобных случаях может быть установлен диагноз как РАКС, так и МДС/МПЗ-Н. Причиной этого является отсутствие четких дифференциально-диагностических критериев (ранее синонимом МДС/МПЗ-Н являлся термин «перекрестный синдром» — «overlap» syndrome). При наличии у больного «синдрома 5q-» в сочетании с тромбоцитозом принято диагностировать вариант МДС с изолированной del(5q). Трудности диагностики миелодиспластических синдромов обусловлены тем, что морфологические изменения не являются специфическими для МДС и встречаются при других патологических состояниях неопухолевой природы. В частности, они характерны для В12 и фолиеводефицитной анемий, возможны при интоксикации тяжелыми металлами, алкогольном поражении печени, различных воспалительных процессах, пароксизмальной ночной гемоглобинурии (ПНГ), наследственной форме болезни Пельгера, СПИДе, а также как следствие проводимой цитостатической терапии. Дифференциальный диагноз гипопластического варианта миелодиспластического синдрома (МДС) проводится с апластической анемией (АА). В первом случае в материале трепанобиопсии костного мозга наблюдается пролиферация трех ростков кроветворения и может выявляться АЛМП. При гипопластическом миелодиспластического синдроме значительно реже происходит увеличение числа тучных клеток. При АА в большинстве случаев определяется только одноростковая (эритроидная) пролиферация без признаков дисплазии гемопоэза. Относительная гиперплазия мегакариоцитов характерна для миелодиспластического синдрома, в то время как при АА наблюдается их выраженная гипоплазия или аплазия. Достоверно различающиеся клинико-лабораторные признаки при первичных и вторичных миелодиспластических синдромов (МДС)
В исследовании I. Lorand-Metze и соавт. в трепанобиоптатах при апластической анемии мегакариоциты, которые авторы выявляли иммуногистохимически, отсутствовали у 12 из 20 больных, в то время как при гипопластическом миелодиспластическом синдроме — у 1 из 21. При апластической анемии также было значительно ниже среднее число мегакариоцитов, а основная часть гемопоэтических клеток была представлена нормобластами и лимфоцитами. При миелодиспластическом синдроме определялась большая неравномерность участков костного мозга, содержащих кроветворные клетки. При иммуногистохимическом исследовании трепанобиоптатов больных с гипопластическим вариантом миелодиспластического синдрома выявлено достоверно большее число CD34+-клеток по сравнению с АА (0,94 и 0,04 % соответственно), а также большее число PCNA-(proliferating cell nuclear antigen — ядерный антиген пролиферирующих клеток) позитивных клеток, чем при АА (43,59 и 14,80 % соответственно). Важным методом исследования, позволяющим диагностировать МДС, является цитогенетическое исследование, которое позволяет обнаружить характерные хромосомные аномалии. Однако имеются работы, указывающие на возможность изменений кариотипа и при АА, например, +6 или t(l;20). Тем не менее в большинстве случаев при АА хромосомные изменения выявляются при длительном течении болезни, либо после иммуносупрессивного лечения (например, моносомия хромосомы 7), либо после трансформации АА в миелодиспластический синдром (МДС) или ОЛ. При первичном обследовании больных АА частота аномалий кариотипа не превышает 4 %. Дифференциальный диагноз при фиброзном варианте миелодиспластического синдрома (МДС) проводится с хроническими миелопро-лиферативными заболеваниями, наиболее часто — с хроническим идиопатическим миелофиброзом (ХИМФ). Последний за редким исключением протекает со спленомегалией, лейкоцитозом со сдвигом формулы крови влево до миелоцитов и тромбоцитозом. В крови и костном мозге отсутствуют многочисленные проявления дисплазии гемопоэтических клеток. При исследовании мазка крови зачастую выявляют нормоцитоз, пойкилоцитоз с каплевидной формой эритроцитов, ядра и фрагменты мегакариоцитов. ХИМФ характеризуется более выраженным фиброзом костного мозга, в том числе коллагеновым. Цитогенетическое исследование иногда позволяет выявить менее характерные для миелодиспластического синдрома изменения: трисомия хромосомы 21, утрата хромосомы 13 или ее длинного плеча. Дополнительным признаком, указывающим на ХИМФ, является повышенный уровень ЩФ гранулоцитов (однако и этот признак нередко обнаруживается при миелодиспластическом синдроме). Необходимость дифференциального диагноза с В12-дефицитной анемией связана с возможной панцитопенией и дисплазией не только эритроидного, но и нейтрофильного и мегакариоцитарного ростков кроветворения при этом заболевании. В типичных случаях В12-дефицитной анемии преобладание мегалобластов среди нормобластов помогает в определении истинной природы заболевания. При «стертой» картине В12-дефицитной анемии диагноз становится очевидным по результатам терапии — ретикулоцитарный криз после 3—5 дней терапии витамином В12. Необходимость дифференциального диагноза с ПНГ обусловлена общей для обоих заболеваний панцитопенией при нормо- или гиперклеточном костном мозге. Отличительным признаком ПНГ является ретикулоцитоз крови, положительные пробы Хема и сахарозная, обнаружение гемосидерина в моче и свободного гемоглобина в сыворотке крови. Иногда возникают трудности в дифференциальном диагнозе ХММЛ и ХМЛ. Как известно, при ХМЛ обнаруживается дисплазия мегакариоцитарного ростка, иногда имеются черты дисплазии и в гранулоцитарном ростке. В связи с наличием лейкоцитоза может быть увеличено абсолютное число моноцитов, в крайне редких случаях в хронической фазе и значительно чаще в фазе акселерации выявляется тромбоцитопения. При исследовании трепанобиоптата в хронической фазе ХМЛ у 40 % больных может обнаруживаться ретикулиновый фиброз, а в фазе акселерации — и коллагеновый фиброз. Однако в большинстве случаев нарастающий лейкоцитоз с характерными изменениями лейкоцитарной формулы и обнаружение t(9;22) и/или слитного гена BCR/ABL при цитогенетическом исследовании позволяют установить правильный диагноз. – Также рекомендуем “Прогноз миелодиспластических синдромов (МДС) – значение кариотипа” Оглавление темы “Диагностика и прогноз миелодиспластических синдромов (МДС)”:
|
Источник
Миелодиспластический синдром – группа гематологических заболеваний, при которых наблюдаются цитопения, диспластические изменения костного мозга и высокий риск возникновения острого лейкоза. Характерные симптомы отсутствуют, выявляются признаки анемии, нейтропении и тромбоцитопении. Диагноз устанавливается с учетом данных лабораторных анализов: полного анализа периферической крови, гистологического и цитологического исследования биоптата и аспирата костного мозга и т. д. Дифференциальный диагноз может представлять значительные затруднения. Лечение – переливание компонентов крови, химиотерапия, иммуносупрессивная терапия, пересадка костного мозга.
Миелодиспластический синдром
Миелодиспластический синдром – группа заболеваний и состояний с нарушениями миелоидного кроветворения и высоким риском развития острого лейкоза. Вероятность развития увеличивается с возрастом, в 80% случаев данный синдром диагностируется у людей старше 60 лет. Мужчины страдают несколько чаще женщин. У детей миелодиспластический синдром практически не встречается. В последние десятилетия гематологи отмечают увеличение заболеваемости среди лиц трудоспособного возраста. Предполагается, что причиной «омоложения» болезни могло стать существенное ухудшение экологической обстановки.
До недавнего времени лечение миелодиспластического синдрома было только симптоматическим. Сегодня специалисты разрабатывают новые методы терапии, однако эффективное лечение этой группы болезней все еще остается одной из самых сложных проблем современной гематологии. Пока прогноз при миелодиспластическом синдроме, в основном, зависит от особенностей течения болезни, наличия или отсутствия осложнений. Лечение осуществляют специалисты в сфере онкологии и гематологии.
Причины и классификация миелодиспластического синдрома
С учетом причин развития различают два типа миелодиспластического синдрома: первичный (идиопатический) и вторичный. Идиопатический вариант выявляется в 80-90% случаев, диагностируется преимущественно у пациентов старше 60 лет. Причины возникновения установить не удается. В числе факторов риска первичного миелодиспластического синдрома – курение, повышенный уровень радиации при выполнении профессиональных обязанностей или проживании в неблагоприятной экологической зоне, частый контакт с бензином, пестицидами и органическими растворителям, некоторые наследственные и врожденные заболевания (нейрофиброматоз, анемия Фанкони, синдром Дауна).
Вторичный вариант миелодиспластического синдрома наблюдается в 10-20% случаев, может возникать в любом возрасте. Причиной развития становится химиотерапия или радиотерапия по поводу какого-то онкологического заболевания. В число лекарственных средств с доказанной способностью вызывать миелодиспластический синдром включают циклофосфан, подофиллотоксины, антрациклины (доксорубицин) и ингибиторы топоизомеразы (иринотекан, топотекан). Вторичный вариант отличается более высокой резистентностью к лечению, более высоким риском развития острого лейкоза и более неблагоприятным прогнозом.
В современной редакции классификации ВОЗ различают следующие типы миелодиспластического синдрома:
- Рефрактерная анемия. Сохраняется более полугода. В анализе крови бласты отсутствуют либо единичные. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная анемия с кольцевыми сидеробластами. Сохраняется более полугода. В анализе крови бласты отсутствуют. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная цитопения с многолинейной дисплазией. В анализе крови тельца Ауэра отсутствуют, бласты отсутствуют либо единичные, выявляются панцитопения и увеличение количества моноцитов. В костном мозге диспластические изменения менее 10% клеток в 1 миелоидной клеточной линии, бластов менее 5%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-1. В анализе крови тельца Ауэра отсутствуют, бластов более 5%, цитопения и увеличение количества моноцитов. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 5-9%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-2. В анализе крови увеличение количества моноцитов, цитопения, бластов 5-19%, могут выявляться тельца Ауэра. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 10-19%, обнаруживаются тельца Ауэра.
- Неклассифицируемый миелодиспластический синдром. В анализе крови цитопения, бласты отсутствуют либо единичные, тельца Ауэра отсутствуют. В костном мозге дисплазия одного мегакариоцитарного либо гранулоцитарного ростка, бластов более 5%, тельца Ауэра отсутствуют.
- Миелодиспластический синдром, ассоциированный с изолированной делецией 5q. В анализе крови анемия, бластов более 5%, возможен тромбоцитоз. В костном мозге более 5% бластов, тельца Ауэра отсутствуют, изолированная делеция 5q.
Симптомы миелодиспластического синдрома
Клиническая симптоматика определяется степенью нарушений миелопоэза. При мягко протекающих расстройствах возможно длительное бессимптомное или стертое течение. Из-за слабой выраженности клинических проявлений некоторые больные не обращаются к врачам, и миелодиспластический синдром обнаруживается во время проведения очередного медицинского осмотра. При преобладании анемии наблюдаются слабость, одышка, плохая переносимость физических нагрузок, бледность кожных покровов, головокружения и обморочные состояния.
При миелодиспластическом синдроме с тромбоцитопенией возникает повышенная кровоточивость, отмечаются десневые и носовые кровотечения, на коже появляются петехии. Возможны подкожные кровоизлияния и меноррагии. Миелодиспластический синдром с выраженными нейтропенией и агранулоцитозом проявляется частыми простудами, стоматитом, синуситом или стрептодермией. В тяжелых случаях возможно развитие пневмонии или сепсиса. Инфекционные заболевания нередко вызываются грибками, вирусами или условно-патогенными микробами. У каждого пятого пациента с миелодиспластическим синдромом выявляется увеличение лимфоузлов, селезенки и печени.
Диагностика миелодиспластического синдрома
Диагноз выставляется с учетом данных лабораторных исследований: анализа периферической крови, биопсии костного мозга с последующим цитологическим исследованием, цитохимических и цитогенетических тестов. В анализе периферической крови больных миелодиспластическим синдромом обычно обнаруживается панцитопения, реже выявляется дву- или одноростковая цитопения. У 90% пациентов наблюдается нормоцитарная либо макроцитарная анемия, у 60% — нейтропения и лейкопения. У большинства больных миелодиспластическим синдромом отмечается тромбоцитопения.
При исследовании костного мозга количество клеток обычно нормальное либо повышенное. Уже на ранних стадиях обнаруживаются признаки дизэритропоэза. Количество бластов зависит от формы миелодиспластического синдрома, может быть нормальным либо увеличенным. В последующем наблюдаются дисгранулоцитопоэз и дисмегакариоцитопоэз. У некоторых больных признаки дисплазии костного мозга выражены очень слабо. В процессе цитогенетического исследования у ¾ больных выявляются хромосомные нарушения. Дифференциальный диагноз миелодиспластического синдрома проводят с В12-дефицитной анемией, фолиево-дефицитной анемией, апластической анемией, острым миелолейкозом и другими острыми лейкозами.
Лечение и прогноз при миелодиспластическом синдроме
Тактика лечения определяется выраженностью клинической симптоматики и лабораторных изменений. При отсутствии явных признаков анемии, геморрагического синдрома и инфекционных осложнений осуществляется наблюдение. При миелодиспластическом синдроме с выраженной анемией, тромбоцитопенией и нейтропенией, а также при высоком риске возникновения острого лейкоза назначают сопроводительную терапию, химиотерапию и иммуносупрессивную терапию. При необходимости осуществляют пересадку костного мозга.
Сопроводительная терапия является самым распространенным методом лечения миелодиспластического синдрома. Предусматривает внутривенные инфузии компонентов крови. При длительном применении может провоцировать повышение уровня железа, влекущее за собой нарушения деятельности жизненно важных органов, поэтому переливания гемокомпонентов производят при одновременном приеме хелаторов (лекарственных средств, связывающих железо и способствующих его выведению).
Иммуносупрессоры эффективны при лечении миелодиспластического синдрома с отсутствием хромосомных аномалий, наличием гена HLA-DR15 и гипоклеточном костном мозге. Химиотерапию применяют при невозможности трансплантации костного мозга. Высокие дозы препаратов используют при трансформации миелодиспластического синдрома в острый лейкоз, а также при рефрактерных анемиях с избытком бластов при нормоклеточном и гиперклеточном костном мозге, низкие – при невозможности пересадки костного мозга. Наряду с перечисленными средствами пациентам назначают гипометилирующие средства (азацитидин). Наиболее надежным способом достижения полноценной длительной ремиссии является трансплантация костного мозга.
Прогноз зависит от типа миелодиспластического синдрома, количества хромосомных аномалий, необходимости в регулярных переливаниях компонентов крови, выраженности клинических проявлений и наличия осложнений. Различают 5 групп риска. Средняя выживаемость больных миелодиспластическим синдромом, входящих в группу с самым низким уровнем риска, составляет более 11 лет; с самым высоким – около 8 месяцев. Вероятность отторжения костного мозга после трансплантации – около 10%.
Источник