Серповидно клеточная анемия наследуется по типу

Поделиться статьей в социальных сетях:
Серповидноклеточная анемия является следствием генных мутаций, на участке, отвечающем за контроль над образованием бета-цепей в сложном белке, в контексте – гемоглобине. Как результат мутации – одна аминокислота в b-глобиновой цепи заменяется. Конкретно: происходит замещение глутаминовой кислоты в 6 позиции на валин.
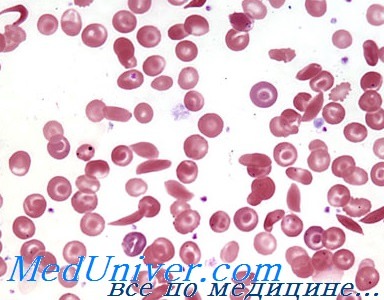
То есть, формула белка теперь неустойчива и на фоне прогрессирующей гипоксии изменяется ее строение. Происходят кристаллизация и полимеризация, образуется измененный гемоглобин HbS. Что становится причиной деструкции формы эритроцитов – они длиннее, истончаются, внешне начинают напоминать серпы.
Наследственная болезнь – серповидноклеточная анемия: что это
Кровь артериального типа оттекает от легких и несет по организму кислород, но на уровне тканей он проникает в клетки всех органов, и это неизбежно приведет к реакции полимеризации белка и появлению эритроцитов с формой полумесяца.
У человека серповидноклеточная анемия обратима только на начальной стадии. Вторичное прохождение легочных капилляров, вновь насыщает кровь кислородом, что возвращает эритроцитам их адекватные формы. Но деструктивные изменения повторяются при прохождении крови через ткани, как результат – строение эритроцитарной мембраны нарушено, проницаемость повышена, ионы калия и йода покидают клетки. На этом моменте кардинальные изменения эритроцита «фиксируются», они меняются необратимо.
 Способность пластической адаптации у серповидных эритроцитов сильно снижена, он уже не может претерпевать обратную деформацию, проходя через капилляры, поэтому закупоривает их. Что приводит к нарушению кровоснабжения разных систем и органов, развивается тканевая гипоксия. Это провоцирует дальнейшее увеличение числа месяцеподобных эритроцитов.
Способность пластической адаптации у серповидных эритроцитов сильно снижена, он уже не может претерпевать обратную деформацию, проходя через капилляры, поэтому закупоривает их. Что приводит к нарушению кровоснабжения разных систем и органов, развивается тканевая гипоксия. Это провоцирует дальнейшее увеличение числа месяцеподобных эритроцитов.
У больных серповидноклеточной анемией эритроцитарная мембрана слишком ломкая и хрупкая, поэтому продолжительность жизни клетки весьма коротка. На фоне этого уменьшается и общее число эритроцитов, появляются локальные сбои в цикле кровообращения на тканевом уровне, закупориваются сосуды, в почках начинает усиленно образовываться эритропоэтин. Это ускоряет процессы эритропоэза в красном веществе костного мозга, за счет чего компенсируется анемичное состояние.
Нужно заметить, что HbF, который состоит и из альфа-цепей, и гамма-цепей, в некоторых эритроцитах по концентрации достигает 10%, при этом не подвержен полимеризирующим реакциям и способен предотвратить деформацию эритроцитов до серповидной формы. Клетки с минимальным содержанием HbF видоизменяются одними из первых, практически сразу же.
Наследование серповидноклеточной анемии
 Как указывалось выше – серповидноклеточная анемия наследуется как генетическое заболевание. Мутация обуславливается изменениями в одном или двух генах, отвечающих за кодирование b-цепей в белке. Такая патология не возникает в организме самостоятельно, а передается от обоих родителей.
Как указывалось выше – серповидноклеточная анемия наследуется как генетическое заболевание. Мутация обуславливается изменениями в одном или двух генах, отвечающих за кодирование b-цепей в белке. Такая патология не возникает в организме самостоятельно, а передается от обоих родителей.
Половые клетки содержат в себе по 23 хромосомы. В момент успешного оплодотворения они сливаются, таким образом появляется зигота, то есть, клетка с новыми качествами. Из нее затем и развивается плод. Сливаются между собой и ядра половых клеток обоих полов, и, по сути, благодаря этому восстанавливается полный хромосомный набор (23 пары). Что и присуще клеткам организма человека. Таким образом новорожденный унаследует генетический материал и от матери, и от отца.
Серповидноклеточная анемия: тип наследования – аутосомно-рецессивный. Чтобы родившийся ребенок был болен, он должен получить мутировавшие гены от обоих родителей. Все зависит о того, какой именно комплект генов унаследовал новорожденный:
- Малыш с диагностированной серповидноклеточной анемией. Но этот вариант будет возможен при соблюдении следующего условия: мать и отец имеют эту патологию или являются ее бессимптомным носителем. Еще одно условие – новорожденный получает по одному «бракованному» гену от каждого. Это называется гомозиготной формой заболевания.
- Опять рождается человек, являющийся бессимптомным носителем. Этот вариант развивается, если малыш получает «в наследство» только один дефективный ген, а второй – нормальный. Это называется гетерозиготным типом заболевания. Как результат – эритроцит содержит приблизительно равное число как гемоглобина типа S, так и типа А. Что помогает поддерживать оптимальную форму и эритроцитарные функции, при условии, что нет никаких отягчающих состояние патологий.
То есть, у человека серповидноклеточная анемия наследуется как не полностью (носитель), так и полностью (болеющий). Других вариантов появления мутаций врачами обнаружено не было. Но точные причины их развития у родителей не были установлены и до сих пор. Предполагают только ряд факторов, приводящих к мутациям, чье прямое действие на организм приведет к искажению генетического клеточного аппарата, провоцируя большой спектр хромосомных патологий.
Серповидная анемия: диагностирование и лечение
Диагностировать и лечить серповидноклеточную патологию может только врач-гематолог. Диагноз не ставят только на основании внешней симптоматики, нужно собрать подробный семейный анамнез, уточнить время и обстоятельства, при которых признаки патологии проявились в первый раз. Но подтвердить диагноз можно только посредством специфических обследований:
Серповидноклеточная анемия в одной из популяций определяется по:
- Традиционному анализу крови.
- Биохимии крови.
- Результатам УЗИ, рентгенографии.
 Эффективных средств лечения, дающих возможность полноценного избавления от этой болезни – не существует. Помочь больному можно только путем предотвращения увеличения количества видоизмененных эритроцитов. Кром того, нужно вовремя купировать внешние признаки болезни.
Эффективных средств лечения, дающих возможность полноценного избавления от этой болезни – не существует. Помочь больному можно только путем предотвращения увеличения количества видоизмененных эритроцитов. Кром того, нужно вовремя купировать внешние признаки болезни.
Принципиальное лечение этой анемии состоит из:
- Здорового образа жизни.
- Лекарств, повышающих показатели белка-гемоглобина и увеличивающих число недеформированных эритроцитов.
- Кислородотерапии.
- Купирования локальных болей.
- Устранения профицита железа.
- Профилактики вирусных инвазий.
Метод, позволяющий установить процентную вероятность наследования патологии – это ПЦР. Исследуется родительский генетический материал и выявляются мутировавшие участки генома. Результатом считается и их наличие/отсутствие, и определение типа и формы заболевания при его наличии – гомозиготная/гетерозиготная анемия.
Источник
Генетика серповидноклеточной анемии. НаследованиеHbS был первым обнаруженным аномальным гемоглобином с высоким клиническим значением. Он возникает вследствие замены единственного нуклеотида, которая изменяет кодон шестой аминокислоты В-глобина глутаминовой кислоты на валин (GAG -> GTG: Glu6Val). Гомозиготность по данной мутации — причина серповидноклеточной анемии, серьезного заболевания, часто встречающегося в некоторых частях света. Болезнь имеет характерное географическое распределение, чаще всего встречается в Экваториальной Африке и реже всего в Средиземноморье, Индии и странах, в которые мигрировали люди из этих регионов. С этой, обычно фатальной в раннем детстве болезнью рождаются около 1 из 600 афроамериканцев, хотя все более частым становится более долгое выживание. Серповидноклеточная анемия — тяжелое аутосомно-рецессивное гемолитическое заболевание, характеризующееся тенденцией эритроцитов принимать выраженно аномальную форму (серпа) в условиях низкого насыщения кислородом. Гетерозиготы, про которых говорят, что они имеют «признак» серповидноклеточно-сти, обычно клинически здоровы, но их эритроциты в условиях очень низкого давления кислорода in vitro принимают форму серпа. Случаи, когда бы это могло происходить in vivo, редки, хотя гетерозиготы имеют риск инфаркта селезенки, особенно при полетах на большой высоте в самолетах с низким давлением в кабине. Гетерозиготное состояние наблюдают приблизительно у 8% афроамериканцев, но в областях, где частота гена высокая (например, в Западной Центральной Африке), вплоть до 25% новорожденных — гетерозиготы. Молекулярная патология HbS – серповидноклеточной анемииОколо 50 лет тому назад Ингрэм обнаружил, что аномалия HbS связана с заменой одной из 146 аминокислот в В-цепи молекулы гемоглобина. Все клинические проявления наличия HbS — последствия этого единственного изменения в гене В-глобина. Это было первой демонстрацией того, что мутация в структурном гене может вызывать замену аминокислоты в соответствующем белке. Поскольку аномалия HbS локализуется в В-цепи, формула HbS может быть записана как а2b2s или, более точно, а2Ab2s.
Гетерозиготы имеют смесь двух типов гемоглобинов (НbА и HbS), обозначаемых а2Аb2А, а2Аbs, а также гибридный тетрамер гемоглобина, обозначаемый как a2AbA,bs. Серповидноклеточность и ее последствияМолекулы гемоглобина, содержащие мутантные субъединицы b-глобина, нормальны по их способности выполнять их главную функцию связывания кислорода (если они не полимеризованы, как указано далее), но в ненасыщенной кислородом крови они растворимы в пять раз меньше по сравнению с нормальным гемоглобином. Относительная нерастворимость дезоксигемоглобина S является физической основой феномена серповидноклеточности. В условиях низкой кислородной напряженности молекулы HbS собираются в форме полимеров, формирующих стержни или волокна, искажающие форму эритроцитов. Эти уродливые эритроциты деформируются хуже, чем в норме, и, в отличие от нормальных красных кровяных клеток, не могут сжиматься, проходя через капилляры, тем самым блокируя ток крови и вызывая локальную ишемию. Происхождение мутаций гемоглобина SУ большинства лиц африканского происхождения нормальный ген b-глобина содержится в пределах фрагмента рестрикции размером в 7,6 килобазы ДНК. В то же время в определенных частях Африки, например в Гане и почти у 70% афроамериканцев, аллель серповидноклеточного глобина часто обнаруживают во фрагменте размером в 13 килобаз. Частая ассоциация серповидноклеточного глобина с 13-килобазовым фрагментом — поразительный пример неравновесного сцепления. В других частях Африки (например, в Кении) мутация серповидноклеточности обычно связана с фрагментом размером в 7,6 килобазы. Эти находки позволяют утверждать, что мутация серповидноклеточности возникла в Западной Африке в хромосоме, которая содержала ген р-глобина во фрагменте длиной 13 килобаз, и что подобная мутация, по крайней мере, один раз, независимо произошла где-то еще. Защита от малярии, обеспечиваемая данной мутацией у гетерозигот, обеспечила ее высокую частоту в областях, пораженных малярией. – Также рекомендуем “Генетика гемоглобинов HbС, Hammersmith. Наследование” Оглавление темы “Генетика гемоглобинопатий”:
|
Источник
Серповидноклеточная анемия: причины, диагностика, лечениеЭтиология и встречаемость серповидноклеточной анемии. Серповидноклеточная анемия (MIM № 603903) — аутосомно-рецессивное заболевание гемоглобина, вызванное миссенс-мутацией гена бета-субъединицы, заменяющей валин на глутаминовую кислоту в 6 положении. Болезнь чаще вызвана гомозиготностью по мутации серповидноклеточности, хотя серповидноклеточную анемию также может вызывать компаундная (составная) гетерозиготность по аллелю серповидноклеточности и аллелям HbC или бета-талассемии. Распространение серповидноклеточной анемии широко изменяется среди популяций в соответствии с прошлым и настоящим распространением малярии. Мутация серповидноклеточности, как оказалось, несколько повышает сопротивляемость малярии, таким образом, давая преимущество выживания гетерозиготным носителям мутации. Патогенез серповидноклеточной анемииГемоглобин формируется из четырех субъединиц: двух а-субъединиц, кодируемых геном ЯВА в хромосоме 16, и двух бета-субъединиц, кодируемых геном ЯВВ в хромосоме 11. Мутация Glu6Val в бета-субъединице уменьшает растворимость ненасыщенного кислородом гемоглобина и вызывает формирование сети жестких волокнистых полимеров, искажающих строение эритроцита, придавая ему форму серпа. Серповидные эритроциты закупоривают капилляры и вызывают инфаркты. Первоначально обогащение кислородом заставляет полимер гемоглобина растворяться, и эритроциты восстанавливают нормальную форму; тем не менее, регулярное нарушение формы приводит к необратимому переходу клеток в серповидную форму, впоследствии такие эритроциты удаляются из кровотока в селезенке. Скорость удаления эритроцитов из кровотока превышает возможность их синтеза в костном мозге, что приводит к гемолитической анемии. Аллельная гетерогенность часто встречается при большинстве менделирующих заболеваний, особенно когда мутантные аллели вызывают снижение функции. Серповидноклеточная анемия — важное исключение из этого правила, поскольку в данном случае единственная специфическая мутация ответственна за уникальные новые свойства HbS. HbC тоже менее растворим, чем HbA, и тоже стремится кристаллизоваться в эритроцитах, уменьшая их деформируемость в капиллярах и вызывая легкий гемолиз, но HbC не формирует полимерные волокна, как HbS. Неудивительно, что другие мутации с новыми функциями, например, мутации в гене FGFR3, вызывающие ахондроплазию, часто имеют аналогичное снижение аллельной гетерогенности, когда фенотип зависит от специфического, уникального изменения функции белка.
Фенотип и развитие серповидноклеточной анемииКлиническая картина у больных серповидноклеточной анемией обычно проявляется в течение первых двух лет жизни анемией, задержкой развития, спленомегалией, регулярными инфекциями и дактилитами (болезненными припухлостями кистей или стоп, вызванными закупоркой капилляров в небольших костях, обнаруженных у приведенной в примере пациентки). Инфаркты вследствие закупорки сосудов происходят во многих тканях, вызывая инсульты мозга, острый кардиальный синдром, почечный папиллярный некроз, инфаркты селезенки, язвы ног, приапизм, асептический некроз костей и снижение зрения. Окклюзия сосудов костей вызывает приступы болей, при отсутствии лечения эти болезненные эпизоды могут продолжаться в течение нескольких дней и даже недель. Функциональная аспления вследствие инфарктов и других недостаточно ясных факторов, предрасполагает к бактериальным инфекциям, например, пневомококковому или сальмонеллезному сепсису и остеомиелиту. Инфекция — основная причина смерти во всех возрастных группах, хотя прогрессирующая почечная и дыхательная недостаточность также нередкие причины смерти на четвертом и пятом десятилетиях жизни. Пациенты также имеют высокий риск развития угрожающей жизни апластической анемии после парвовирусной инфекции, поскольку парвовирусы вызывают временное прекращение образования эритроцитов. Гетерозиготные носители мутации («признака» серповидноклеточности) не имеют анемии и обычно клинически здоровы. Однако в условиях серьезной гипоксии, например, при восхождении в горы, эритроциты пациентов с «признаком» серповидноклеточности могут принимать форму серпа, вызывая симптомы, подобные наблюдаемым при серповидноклеточной анемии. Особенности фенотипических проявлений серповидноклеточной анемии: Лечение серповидноклеточной анемииКонкретному больному серповидноклеточной анемией дать точный прогноз тяжести течения болезни невозможно. Хотя молекулярная основа болезни стала известной раньше других моногенных заболеваний, лечение остается только симптоматическим. Никакой специфический терапии, предохраняющей от процесса образования серповидных эритроцитов, не найдено. Существенно снижает тяжесть болезни персистенция HbE Исследуется несколько фармакологических препаратов, нацеленных на увеличение концентрации HbF, в этих целях одобрено использование гидрокси-мочевины. Хотя генотерапия имеет шанс улучшить или излечить эту болезнь, эффективная пересадка гена b-глобина не достигнута. Пересадка костного мозга остается единственным доступным в настоящее время лечением, способным помочь при серповидноклеточной анемии. Из-за 11% смертности, вызванной сепсисом в первые 6 мес жизни, большинство штатов в США проводит неонатальный скрининг на серповидноклеточную анемию с целью проведения профилактики антибиотиками, продолжающейся до 5-летнего возраста. Риски наследования серповидноклеточной анемииПоскольку серповидноклеточная анемия — аутосомно-рецессивное заболевание, будущие сибсы больного ребенка имеют 25% риск серповидноклеточной анемии и 50% риск носительства серповидноклеточности. Используя ДНК плода, полученную при БВХ или амниоцентезе, можно провести пренатальную диагностику обнаружением мутации. Пример серповидноклеточной анемии. Второй раз за полгода семейная пара карибского происхождения обратилась со своей 24-месячной дочерью в отделение неотложной помощи, поскольку девочка не может стоять. В анамнезе отсутствуют повышение температуры, инфекция или травма, и в остальном медицинская история ничем не примечательна; данные предыдущих осмотров соответствовали норме, за исключением низкого уровня гемоглобина и слегка увеличенной селезенки. При текущем осмотре патологии не найдено, за исключением пальпируемого края селезенки и отека стоп. Стопы болезненны при пальпации, и девочка не хотела вставать на ноги. Оба родителя имели сибсов, умерших в детстве от инфекций, и других сибсов, вероятно, имевших серповидноклеточную анемию. С учетом анамнеза и повторного болезненного увеличения стоп врач проверил ребенка на наличие серповидноклеточной анемии методом электрофореза гемоглобина. Результат этого теста подтвердил наличие HbS. – Также рекомендуем “Болезнь Тея-Сакса: причины, диагностика, лечение” Оглавление темы “Врожденные болезни”:
|
Источник