В настоящее время известно много редких форм гемоглобина

Раздел I. Молекулярная генетика
| Таблица генетического кода (по С.Е. Бреслеру, 1963) | |
| Аминокислота | Предполагаемая структура кодирующего триплета информационной РНК |
| Глицин | ГУГ |
| Аргинин | ГУЦ |
| Валин | УУГ |
| Гистидин | АУЦ |
| Изолейцин | УУА |
| Цистеин | ГУУ |
| Лейцин | УАУ, УУЦ, УГУ |
| Пролин | ЦУЦ, УЦЦ |
| Фенилаланин | УУУ |
| Аланин | ЦУГ |
| Метионин | УГА |
| Тирозин | АУУ |
| Триптофан | УГГ |
| Серин | ЦУУ УЦУ |
| Треонин | УЦА |
| Лизин | АУА |
| Глутаминовая кислота | АУГ |
| Глутамин | УЦГ |
| Аспарагиновая кислота | ГУА |
| Аспарагин | ЦУА, УАА |
Исследование роли ДНК и РНК в передаче наследственных свойств и установление схемы биосинтеза белка и его регуляции позволяет анализировать наследственность
на молекулярном урввне. Ген в конечном итоге – это участок ДНК. Малейшее изменение структуры ДНК ведет к изменениям белка-фермента, что в свою очередь изменяет
цепь биохимических реакций в организме, определяющих тот или иной признак, или серию признаков. Таким образом, признак зависит от характера биохимической реакции,
реакция управляется белком-ферментом, строение белка зашифровано в ДНК посредством специфического чередования азотистых оснований в цепи. Во время синтеза белка
чередование азотистых оснований ДНК копирует информационная РНК (транскрипция). Затем на рибосоме транспортные РНК расшифровывают эту “запись”, расставляя
поочередно аминокислоты соответственно триплетам азотистых оснований (трансляция).
Зная структуру белка-фермента, можно расшифровать строение ДНК, и наоборот,
зная изменения в ДНК, можно предусмотреть изменения в структуре белка. Например, если цепь ДНК, кодирующая какой-то полипептид, начинается: аденин – аденин –
цитозин – гуанин – аденин – тимин (ААЦГАТ), то информационная РНК снимает копию следующим образом: урацил – урацил – гуанин – цитозин – урацил – аденин (УУГЦУА).
Сопоставляя с таблицей генетического кода, можно увидеть, что первым трем азотистым основаниям информационной РНК соответствует аминокислота валин, следующим трем
основаниям – аспарагин. Следовательно, полипептид будет начинаться с аминокислот валин – аспарагин.
Предлагаемые задачи рассчитаны на расшифровку структуры белка по известным изменениям в ДНК и обратный анализ с помощью таблицы кодирования аминокислот.
Задачи
- Полипептид состоит из следующих друг за другом расположенных аминокислот: валин – аланин – глицин – лизин – триптофан – валин – серии – глутаминовая
кислота. Определите структуру участка ДНК, кодирующего вышеуказанный полипептид
[показать] - Полипептид состоит из следующих аминокислот: аланин – цистеин – гистидин – лейцин – метионин – тирозин. Определите структуру участка ДНК, кодирующего эту полипептидную цепь.
- Аспарагин – глицин – фенилаланин – пролин – треонин – аминокислоты последовательно составляют полипептид. Определите структуру участка ДНК, кодирующего данный полипептид.
- Первые 10 аминокислот в цепи В инсулина: фенилаланин – валин – аспарагиновая кислота – глутамин – гистидин – лейцин – цистеин – глицин – серии – гистидин.
Определите структуру участка ДНК, кодирующего эту часть цепи инсулина
[показать] - Начальный участок цепи A инсулина представлен следующими пятью аминокислотами: глицин – изолейцин – валин – глутамин – глутамин.
Определите участок ДНК, кодирующий эту часть цепи инсулина
[показать] - В цепи рибонуклеазы поджелудочной железы один из полипептидов имеет следующие аминокислоты: лизин – аспарагиновая кислота – глицин – треонин – аспарагиновая
кислота – глутаминовая кислота – цистеин. Определите информационную РНК, управляющую синтезом указанного полипептида. - Одна из цепей рибонуклеазы поджелудочной железы состоит из следующих 14 аминокислот: глутамин – глицин – аспарагиновая кислота – пролин – тирозин – валин –
пролин – валин – гистидин – фенилаланин – аспарагин – аланин – серии – валин. Определите структуру участка ДНК, кодирующего эту часть цепи рибонуклеазы
[показать] - Одна из цепей глюкагона имеет следующий порядок аминокислот: треонин – серии – аспарагин – тирозин – серии – лизин – тирозин. Определите строение участка ДНК, кодирующего эту часть цепи глюкагоиа.
- Участок молекулы ДНК, кодирующий часть полипептида, имеет следующее строение: АЦЦАТТАЦЦАТГАА. Определите последовательность аминокислот в полипептиде
[показать] - При синдроме Фанкони (нарушение образования костной ткани) у больного с мочой выделяются аминокислоты, которым соответствуют следующие триплеты
информационной РНК: АУА, ГУЦ, АУГ, УЦА, УУГ, УАУ, ГУУ, АУУ. Определите, выделение каких аминокислот с мочой характерно для синдрома Фанкони
[показать] - У человека, больного цистинурией (содержание в моче большего, чем в норме, числа аминокислот) с мочой выделяются аминокислоты, которым соответствуют
следующие триплеты информационной РНК: ЦУУ, ГУУ, ЦУГ, ГУГ, УЦГ, ГУЦ, АУА. У здорового человека в моче обнаруживается аланин – серии – глутаминовая кислота и
глицин. Выделение каких аминокислот с мочой характерно для больных цистинурией? Напишите триплеты, соответствующие аминокислотам, имеющимся в моче здорового
человека
[показать] - Как изменится структура белка, если из кодирующего его участка ДНК – ГАТАЦТТАТАААГАЦ удалить пятый и тринадцатый (слева) нуклеотиды?
[показать] - Какие изменения произойдут в строении белка, если в кодирующем его участке ДНК – ТААЦАГАГГАЦТААГ между 10 и 11 нуклеотидом включен цитозин, между 13 и 14 –
тимин, а на конце рядом с гуанином прибавился еще один гуанин? - Участок молекулы ДНК, кодирующий полипептид, имеет в норме следующий порядок азотистых оснований: АААААЦЦАТАГАГАГААГТАА. Во время репликации третий слева
аденин выпал из цепи. Определите структуру полипептидной цепи, кодируемой данным участком ДНК, в норме и после выпадения аденина. - Участок цепи белка вируса табачной мозаики состоит из следующих аминокислот: серии – глицин – серии – изолейцин – треонин – пролин – серин. В результате
воздействия на информационную РНК азотистой кислотой цитозин РНК превращается в гуанин. Определите изменения в строении белка вируса после воздействия на РНК
азотистой кислотой
[показать] - Четвертый пептид в нормальном гемоглобине (гемоглобин А) состоит из следующих аминокислот: валин – гистидин – лейцин – треонин – пролин – глутаминовая кислота
– глутаминовая кислота – лизин.- У больного с симптомом спленомегалии при умеренной анемии обнаружили следующий состав 4-го пептида; валин – гистидин – лейцин – треонин – пролин – лизин – глутаминовая кислота – лизин. Определите изменения, произошедшие в ДНК, кодирующей 4-й пептид гемоглобина, после мутации.
- У больного серповидноклеточной анемией состав аминокислот 4-го пептида гемоглобина следующий: валин – гистидин – лейцин – треонин – пролин – валин – глутаминовая
кислота – лизин. Определите изменения в участке ДНК, кодирующем 4-й пептид гемоглобина, приведшие к заболеванию.
- В четвертом пептиде нормального гемоглобина А 6-я и 7-я позиция представлена двумя одинаковыми аминокислотами: глутаминовая кислота – глутаминовая кислота.
У других форм гемоглобина произошли следующие замещения:Форма гемоглобина Аминокислота в позиции 6 7 S Валин Глутаминовая кислота G Лизин Глутаминовая кислота G Глутаминовая кислота Глицин Джоржтауп Глутаминовая кислота Лизин Определите структуру участков ДНК, кодирующих 6-ю и 7-ю позицию четвертого пептида для всех пяти форм гемоглобина.
- В настоящее время известно много редких форм гемоглобина, у которых в результате мутаций произошло замещение той или иной аминокислоты в α-цепи.
- У гемоглобина Торонто 5-я аминокислота аланин заменена аспарагином, у гемоглобина Париж 6-я аминокислота аланин заменена аспарагином.
Определите участок ДНК, кодирующий 5-ю и 6-ю аминокислоты α-цепи, для нормального гемоглобина А и для гемоглобинов Торонто и Париж
[показать] - У гемоглобина Интерлакен-Оксфорд 15-я аминокислота глицин заменена аспарагином, у гемоглобина J 16-я аминокислота лейцин заменена глутамином.
Определите участок ДНК, кодирующий 15-ю и 16-ю аминокислоты α-цепи, у нормального гемоглобина и у обоих измененных
[показать]
- У гемоглобина Торонто 5-я аминокислота аланин заменена аспарагином, у гемоглобина Париж 6-я аминокислота аланин заменена аспарагином.
- Известно 26 форм гемоглобина, у которых произошла замена той или иной аминокислоты в β-цепи (В. П. Эфроимсон, 1968). В таблице приведены некоторые
замещения. Напишите изменения в триплетах ДНК, приведших к изменениям гемоглобина.Форма гемоглобина Порядковый номер аминокислоты в цепи Аминокислотные замещения Токучи 2 Гистидин – тирозин Кушатта 22 Глутамин – аланин Айбадан 46 Глицин – глутамин Цюрих 63 Гистидин – аргинин Милуоки 67 Валин – глутамин Ибадан 87 Треонин – лизин Балтимор 95 Лизин – глутамин Кельн 98 Валин – метионин О-Аравия 121 Глутамин – лизин Хоп 136 Глицин – аспарагин Кенвуд 143 Гистидин – аспарагин - В цепи А инсулина лошади аминокислоты в позиции 6-11 имеют следующий состав: цистеин – цистеин – треонин – глицин – изолейцин – цистеин. У быка в этой цепи
8-ю позицию занимает аланин, 9-ю – серин, 10-ю – валин. Определите строение участка ДНК, кодирующего эту часть цепи инсулина, у лошади и быка. - Начальный участок цепи В инсулина представлен следующими 10 аминокислотами: фенилаланин – валин – аспарагиновая кислота – глутамин – гистидин – лейцин –
цистеин – глицин – серин – гистидин. Определите количественные соотношения аденин + тимин, гуанин + цитозин в цепи ДНК, кодирующей этот участок инсулина
[показать] - Инсулин состоит из А и В цепи, включающих 51 аминокислоту. Однако состав инсулина лошади, быка и барана несколько отличен. Число различных аминокислот в
Аминокислота Число аминокислот а инсулине животных бык баран лошадь Глицин 4 5 5 Валин 5 5 4 Изолейцин 1 1 2 Лейцин 6 6 6 Фенилаланин 3 3 3 Тирозин 5 5 5 Серин 3 2 2 Треонин 1 1 2 Лизин 1 1 1 Аргинин 1 1 1 Гистидин 2 2 2 Цистеин 6 6 6 Пролин 1 1 1 Аланин 3 3 2 Глутамин 6 6 6 Аспарагиновая кислота 3 3 3 Определите количественные отношения аденин + тимин и гуанин + цитозин в цепи ДНК, кодирующей инсулин, у трех видов животных.
- Четвертый пептид гемоглобинов включает 8 аминокислот. Количественный состав их в различных формах гемоглобина приведен ниже.
Аминокислота Число аминокислот в пептиде гемоглобина А S C G Джоржтаун Валин 1 2 1 1 1 Гистидин 1 1 1 1 1 Лейцин 1 1 1 1 1 Треонин 1 1 1 1 1 Пролин 1 1 1 1 1 Глутаминовая кислота 2 1 1 1 1 Лизин 1 1 2 1 2 Глицин 1 Определите количественные соотношения аденин + тимин и гуанин + цитозин в участке ДНК, кодирующем 4-й полипептид; для пяти форм гемоглобина.
- Рибонуклеаза поджелудочной железы быка имеет следующий количественный состав аминокислот:
Лизин 10 Тирозин 5 Глутамин 6 Цистеин 8 Аргинин 4 Пролин 4 Изолейцин 3 Аспарагиновая кислота 12 Аланин 12 Треонин 10 Лейцин 2 Серин 15 Валин 8 Глутаминовая кислота 7 Метионин 4 Фенилаланин 3 Аспарагин 4 Гистидин 4 Глицин 3 Определите количественные соотношения аденин + тимин и гуанин + цитозин в участке цепи ДНК, кодирующем рибонуклеазу.
- Исследования показали, что 34% общего числа нуклеотидов данной информационной РНК приходится на гуанин, 18%-на урацил, 28% – на цитозин и 20% – на аденин.
Определите процентный состав азотистых оснований двухцепочечной ДНК, слепком с которой является вышеуказанная информационная РНК. - Известно, что расстояние между двумя соседними нуклеотидами в спирализованной молекуле ДИК, измеренной вдоль оси спирали, составляет 34 x 1011 м.
Какую длину имеют гены, определяющие молекулу нормального гемоглобина, включающего 287 аминокислот? - Какую длину имеет молекула ДНК, кодирующая инсулин быка, если известно, что молекула инсулина быка имеет 51 аминокислоту, а расстояние между двумя
соседними нуклеотидами в ДНК равно 34 x 1011 м?
Виртуальные консультации
На нашем форуме вы можете задать вопросы о проблемах своего здоровья, получить
поддержку и бесплатную профессиональную рекомендацию специалиста, найти новых знакомых и
поговорить на волнующие вас темы. Это позволит вам сделать собственный выбор на основании
полученных фактов.

Обратите внимание! Диагностика и лечение виртуально не проводятся!
Обсуждаются только возможные пути сохранения вашего здоровья.
Подробнее см. Правила форума

Последние сообщения
Реальные консультации
Реальный консультативный прием ограничен.
Ранее обращавшиеся пациенты могут найти меня по известным им реквизитам.
Заметки на полях

Нажми на картинку –
узнай подробности!
Новости сайта
Ссылки на внешние страницы
20.05.12
Уважаемые пользователи!
Просьба сообщать о неработающих ссылках на внешние страницы, включая ссылки, не выводящие прямо на нужный материал,
запрашивающие оплату, требующие личные данные и т.д. Для оперативности вы можете сделать это через форму отзыва, размещенную на каждой странице.
Ссылки будут заменены на рабочие или удалены.
Тема от 05.09.08 актуальна!
Остался неоцифрованным 3-й том МКБ. Желающие оказать помощь могут заявить об этом на
нашем форуме
05.09.08
В настоящее время на сайте готовится полная
HTML-версия МКБ-10 – Международной классификации болезней, 10-я редакция.
Желающие принять участие могут заявить об этом на нашем форуме
25.04.08
Уведомления об изменениях на сайте можно получить через
раздел форума “Компас здоровья” – Библиотека сайта “Островок здоровья”
Источник
Вариант 2
Решите задачи 1 – 15:
1. В настоящее время известно много редких форм гемоглобина, у которых в результате мутаций произошло замещение той или иной аминокислоты в α-цепи. В α-цепи нормального гемоглобина А пятая и шестая аминокислоты представлены аланином. У гемоглобина Торонто пятая аминокислота аланин замещена аспарагином, у гемоглобина Париж шестая аминокислота аланин заменена аспарагином. Определите участок ДНК, кодирующий пятую и шестую аминокислоты α-цепи, для нормального гемоглобина А и для гемоглобинов Торонто и Париж.
2. Какую длину имеет часть молекулы ДНК, кодирующая инсулин быка, если известно, что молекула инсулина белка имеет 51 аминокислоту, а расстояние между двумя соседними нуклеотидами в ДНК равно 34×10-11 м.
3. Известно, что определенный ген эукариотической клетки содержит 4 интрона (два по 24 нуклеотида и два по 36 нуклеотидов) и 3 экзона (два по 120 нуклеотидов и один 96 нуклеотидов). Определите: количество нуклеотидов в мРНК; количество кодонов в мРНК; количество аминокислот в полипептидной цепи; количество тРНК, участвующих в трансляции.
4. Семейная гиперхолистеринемия наследуется доминантно через аутосомы. У гетерозигот это заболевание выражается в высоком содержании холестерина в крови, у гомозигот, кроме того, развиваются ксантомы (доброкачественная опухоль) кожи и сухожилий, атеросклероз.
Определите вероятность рождения больных детей в семье, где оба родителя имеют только высокое содержание холестерина в крови.
5. Нечувствительность к запахам (аносмия) детерминирована редким аутосомным доминантным геном. В семье, где один из супругов имеет аносмию, а другой чувствителен к запахам, родились разнозиготные близнецы. Определите возможные генотипы и фенотипы детей.
6. У человека брахидактилия (укорочение пальцев) – доминантный признак, а альбинизм – рецессивный. Какова вероятность рождения ребенка с двумя аномалиями у гетерозиготных по обоим признакам родителей?
7. У человека имеется две формы глухонемоты, которые определяются рецессивными аутосомными не сцепленными генами (то есть находящиеся в разных парах хромосом).
Какова вероятность рождения детей глухонемыми в семье, где мать и отец страдают одной и той же формой глухонемоты, а по другой форме глухонемоты они гетерозиготны?
8. Карий цвет глаз, праворукость и синдактилия (срощенные пальцы) – доминантные – доминантные не сцепленные между собой признаки. Какова вероятность рождения голубоглазых правшей, больных синдактилией, если супруги являются тригетерозиготами по этим генам?
9. У мальчика I группа крови, а у его сестры IV. Определите группы крови родителей.
10. Ангиоматоз сетчатой оболочки наследуется как доминантный аутосомный признак с пенетрантностью 50%.
Определите вероятность заболевания детей в семье, где оба родителя являются гетерозиготными носителями ангиоматоза.
11. У человека имеется несколько форм наследственной близорукости. Умеренная форма (от –2,0 до –4,0) и высокая (выше –5,0) передаются как аутосомные доминантные признаки, не сцепленные между собой. В семье, где мать была близорукой, а отец имел нормальное зрение, родились двое детей: дочь и сын. У дочери оказалась умеренная форма близорукости, а у сына высокая. Какова вероятность рождения следующего ребенка в семье без аномалий, если известно, что у матери близорукостью страдал только один из родителей. Следует иметь в виду, что у людей, имеющих гены обоих форм близорукости, проявляется только одна – высокая.
12. Перепончатопалость передается через Y-хромосому. Определите возможные фенотипы детей от брака перепончатопалого мужчины и нормальной женщины.
13. У человека цветовая слепота обусловлена рецессивным геном, сцепленным с Х-хромосомой. Нормальное зрение определяется доминантным аллелем этого гена. От брака родителей с нормальным зрением родился ребенок с цветовой слепотой. Определить генотипы всех членов семьи.
14. Мужчина получил от отца доминантный ген резус-положительности Rh+ и рецессивный ген, обусловливающий нормальную форму эритроцитов (е), а от матери – рецессивный ген резус-отрицательности rh- и доминантный ген Е, обусловливающий образование эллиптических эритроцитов. Его супруга – резус-отрицательна, с нормальными эритроцитами. Какова вероятность (в процентах), что ребенок будет по этим признакам похож на отца?
15. Доминантные гены катаракты и эллиптоцитоза расположены в первой аутосоме. Определить вероятные фенотипы и генотипы детей от брака здоровой женщины и дигетерозиготного мужчины. Кроссинговер отсутствует.
Таблица генетического кода
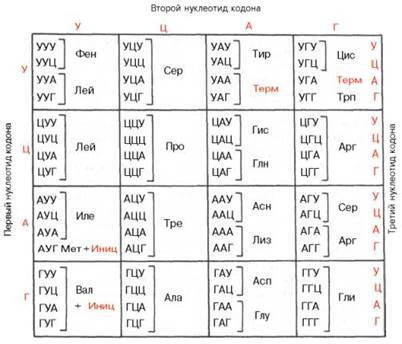
Источник
Гемоглобинопатия – это наследственные заболевания с единой проблемой – образованием аномальной формы гемоглобина, например, серповидноклеточная анемия S и талассемия.
Гемоглобинопатии носят эндемический характер – они возникают в определенном географическом районе, например, в Средиземноморье, Африке, Юго–Восточной Азии. В нашей стране они тоже встречаются.
Что такое гемоглобинопатия
Гемоглобинопатии – это заболевания, вызванные выработкой и присутствием аномальной формы гемоглобина.
Гемоглобин состоит из гема (частей, содержащих железо) и глобина (частей белка, состоящих из аминокислотных цепей). Молекулы гемоглобина (Hb или Hgb) находятся в красных кровяных тельцах. Их задача – связывать кислород в легких и передавать его тканям и органам, где они его выделяют.
Строение гемоглобина
Существует несколько типов цепей глобина: альфа, бета, дельта и гамма.
Типы нормального гемоглобина:
- A – HbA: составляет около 95-98% от общего гемоглобина у взрослых людей. Он содержит 2 альфа (α) цепи и две бета (β) цепи.
- A2 – HbA2: составляет около 2-3% от общего гемоглобина. Он содержит 2 цепи альфа (α) и две цепи дельта (δ).
- F (HbF): составляет около 2% от общего гемоглобина взрослого человека. Он содержит 2 альфа (α) цепи и две гамма (γ) цепи. Этот гемоглобин в основном вырабатывается у плода, его производство значительно снижается вскоре после рождения и достигает уровня взрослого человека в течение 1-2 лет.
К гемоглобинопатиям относятся: структурные варианты гемоглобина, гемоглобин S, серповидноклеточная анемия, гемоглобинопатия C, гемоглобинопатия E, талассемия, гемоглобин Бартс, наследственная персистенция гемоглобина плода.
Причины развития гемоглобинопатии
Гемоглобинопатии возникают в случае генетических изменений генов глобина, которые приводят к изменению аминокислот, составляющих белок глобина. Эти изменения влияют на:
- структуру гемоглобина, например, гемоглобин S, который вызывает серповидно-клеточную анемию;
- его поведение;
- количество продуцируемого вещества (талассемия);
- стабильность.
Серповидно-клеточная анемия
Существует четыре гена, кодирующих цепь альфа-глобина, и два гена, кодирующих цепь бета-глобина. Наиболее частым заболеванием, связанным с изменением альфа-цепи, является альфа-талассемия. Его тяжесть зависит от количества пораженных генов.
Талассемия характеризуется снижением продукции одной из цепей глобина, дисбалансом между альфа- и бета-цепями в гемоглобине A (альфа-талассемия) или увеличением малых форм, таких как Hb A2 или Hb F (бета-талассемия).
Изменения бета-цепей гемоглобина являются врожденными, аутосомно-рецессивными. Это означает, что больной человек должен иметь две дефектные копии гена, каждая от одного из родителей. Если один ген нормален, а другой дефектен, человек гетерозиготен, и мы называем его носителем. Аномальный ген может быть передан любому из потомков. Если рассматриваемый человек является гетерозиготным носителем, он может не иметь никаких симптомов и носительство не влияет на его здоровье.
Если происходят две модификации одного и того же бета-гена, человек гомозиготен по этому гену. Его организм может производить дефектный гемоглобин – возникает гемоглобинопатия с симптомами и потенциальными осложнениями. Степень тяжести зависит от генетической мутации и варьируется от случая к случаю. Копию гена можно передать потомству.
Если два аномальных бета-гена являются врожденными, человек является двойным, смешанным гетерозиготным. У него будут симптомы одной или обеих гемоглобинопатий. Один из аномальных бета-генов будет передаваться каждому из потомков.
Были идентифицированы сотни гемоглобинопатий в бета-цепях. Хотя лишь некоторые из них являются общими и клинически значимыми.
Клинические признаки и симптомы
Признаки и симптомы различаются по типу гемоглобинопатии и возможному сочетанию нескольких гемоглобинопатий. Некоторые приводят к усилению распада эритроцитов (гемолизу), уменьшению их общего количества и развитию анемии.
Клинические признаки включают:
- слабость, утомляемость;
- недостаток энергии;
- желтуха;
- бледность кожи.
Утомляемость
К серьезным клиническим признакам относятся:
- приступы сильной боли;
- удушье;
- увеличение селезенки;
- нарушения роста у детей;
- боль в верхней части живота (вызванная желчными камнями).
Удушье
Общие гемоглобинопатии
Красные кровяные тельца, содержащие аномальный гемоглобин, могут не переносить кислород достаточно эффективно. Они могут разрушаться раньше (чем в здоровых клетках крови) и развиваться гемолитическая анемия. Выявлены сотни гемоглобинопатий, но лишь некоторые из них являются общими и клинически значимыми.
Одной из наиболее распространенных гемоглобинопатий является серповидно-клеточная анемия с присутствием гемоглобина S. Это приводит к изменению формы – серповидно-клеточной – эритроцитов и снижению их выживаемости. Гемоглобин С может вызвать легкую гемолитическую анемию. Гемоглобин E обычно не приводит к развитию каких-либо или только очень легких клинических симптомов.
- Талассемия: самая распространенная генетическая аномалия в мире. Она часто встречается в Средиземноморье, на Ближнем Востоке и в Юго-Восточной Азии. Более легкая форма талассемии также встречается, например, у людей, родившихся в Чехии.
- Гемоглобин S: это основной гемоглобин людей с серповидно-клеточной анемией. В среднем эта мутация есть в одном из двух бета-генов у 8% американцев и африканцев. Возникновение этих мутаций в наших широтах встречаеся редко. Пациенты с заболеванием HbS имеют две аномальные цепи бета (b s) и две нормальные цепи альфа (a). Когда эритроциты, содержащие гемоглобин S, подвергаются действию пониженного количества кислорода (как это может быть в случае повышенной физической нагрузки или инфекционного заболевания легких), они деформируются, принимая форму полумесяца. Серповидные эритроциты могут блокировать периферические кровеносные сосуды и вызывать нарушения кровотока и боль. У них пониженная способность переносить кислород и более короткий срок жизни. Одна копия б не вызывает клинических проявлений, если не сочетается с другой мутацией гемоглобина, такой как HbC (b C) или бета-талассемия.
- Гемоглобин C: около 25% жителей Западной Африки и 2-3% афроамериканцев гетерозиготны по гемоглобину C (у них есть одна копия B C). Но заболевают только гомозиготные люди с обоими дефектными генами (b C). Обычные симптомы – легкая гемолитическая анемия с небольшим или средним увеличением селезенки.
- Гемоглобин E: вторая по распространенности гемоглобинопатия в мире с изменением бета-цепей. Патология очень часто встречается в Юго-Восточной Азии, особенно в Камбодже, Лаосе и Таиланде, а также частично в Северо-Восточной Азии. Есть случаи и в нашей стране. Люди с гомозиготным Hb E (две копии b E) обычно имеют легкую гемолитическую анемию, микроциты (маленькие красные кровяные тельца) и слегка увеличенную селезенку. Одна копия гемоглобина E не вызывает клинических признаков, если не сочетается с другой мутацией, такой как одна из бета-талассемии.
Талассемия
Необычные гемоглобинопатии
Существует ряд гемоглобинопатий, некоторые из которых не проявляются – они не вызывают никаких клинических признаков или симптомов. Другие, в свою очередь, влияют на функциональность и / или стабильность молекулы гемоглобина. Примерами являются гемоглобин D, гемоглобин G, гемоглобин J, гемоглобин M и гемоглобин Constant Spring. Мутации в гене альфа-цепи глобина приводят к образованию аномально длинных альфа (а) цепей, которые вызывают нестабильность в молекуле гемоглобина.
Другие примеры мутаций бета-цепи:
- Гемоглобин F: Hb F в основном вырабатывается в организме будущего ребенка (плода), и его функция заключается в переносе кислорода в среде с низким содержанием кислорода. Продукция гемоглобина F снижается сразу после рождения и стабилизируется на уровне взрослого человека до 1-2 лет. Гемоглобин F может быть повышен при некоторых врожденных заболеваниях. При бета-талассемии его уровень может быть нормальным или повышенным, но часто повышен при серповидно-клеточной анемии и сочетании серповидно-клеточной анемии с бета-талассемией. Пациенты с серповидно-клеточной анемией и повышенным Hb F часто имеют более легкое течение болезни, поскольку Hb F предотвращает серповидное движение красных кровяных телец. Уровни Hb F повышены в редком состоянии, называемом врожденным постоянством выработки гемоглобина плода (HPFH). Люди с повышенным уровнем гемоглобина F не имеют клинических признаков. HPFH вызывается разными генными мутациями у разных этнических групп. Hb F также может быть повышен при некоторых приобретенных состояниях, влияющих на выработку красных кровяных телец. Например, лейкемия и миелопролиферативные заболевания часто сопровождаются небольшим повышением уровня гемоглобина F.
- Гемоглобин H: HbH – это аномальный гемоглобин, который возникает в некоторых случаях альфа-талассемии. Его образование является ответом на фундаментальный недостаток альфа (а) цепей. Hb H состоит из четырех цепей бета (b) глобина. Хотя каждая из цепей бета-глобина нормальна, комплекс из четырех цепей бета нормально не функционирует. Обладает повышенным сродством к кислороду, плохо выделяет кислород клеткам тканей. Присутствие гемоглобина H также связано со значительным распадом эритроцитов (гемолизом), который возникает в результате осаждения нестабильного гемоглобина внутри красных кровяных телец.
- Hemoglobin Barts: Hb Barts вырабатывается в организме будущего ребенка с альфа-талассемией при условии, что все четыре гена, отвечающие за производство гемоглобина альфа, отсутствуют. Таким образом, не может образовываться гемоглобин HbA, HbA 2 и HbF. Гемоглобин Бартс состоит из четырех гамма (g) цепей и имеет высокое сродство к кислороду. Это состояние несовместимо с жизнью и обычно приводит к внутриутробной гибели плода.
Некоторые люди могут унаследовать два гена с разными мутациями, каждый от одного из родителей. Таких людей называют двойными или смешанными гетерозиготами.
Обследование: лабораторные тесты
Исследование на гемоглобинопатию проводится в следующих случаях:
- Выявление гемоглобинопатий у бессимптомных родителей больных детей.
- Выявление гемоглобинопатий у пациента с необъяснимой анемией, микроцитозом и / или гипохромией. Анализ может быть выполнен как часть теста на анемию.
- Скрининг на гемоглобинопатии у новорожденных – только в США и некоторых регионах с повышенной заболеваемостью.
- Пренатальный скрининг проводится в некоторых регионах с высокой частотой гемоглобинопатий (особенно в Африке).
На результаты тестов на гемоглобинопатию может повлиять переливание крови. Поэтому после переливания крови, прежде чем сдать анализ, пациенту следует подождать несколько месяцев. Тем не менее пациентам с серповидно-клеточной анемией после переливания крови рекомендуется сдать анализ крови, чтобы увидеть, достаточно ли гемоглобина в крови, и снизить риск повреждения организма серповидными эритроцитами.
Обследование гемоглобинопатий основано на обнаружении и оценке «нормальности» эритроцитов и гемоглобина в эритроцитах, а также на исследовании конкретной мутации гена. Каждый из тестов является частью головоломки, предоставляющей важную информацию о том, какая гемоглобинопатия присутствует. Для проверки гемоглобинопатии используются следующие тесты:
- Анализ крови. Анализ крови дает быструю информацию о клетках, циркулирующих в крови. Помимо прочего, результаты анализа крови показывают, сколько красных кровяных телец (эритроцитов) содержится в крови, какого они размера и формы, а также сколько там гемоглобина. Размер эритроцита определяет средний объем эритроцитов (MCV). Обнаружение пониженного MCV (микроцитоз, наличие небольших эритроцитов) часто сначала указывает на возникновение талассемии. Если MCV низкий и дефицит железа исключен, пациенты могут быть носителями талассемии или гемоглобинопатии, которые также вызывают микроцитоз (например, HbE).
- Анализ ДНК. Этот анализ используется для скрининга мутаций и делеций в альфа- и бета-областях глобиновых генов. Иногда обследуются все члены семьи. Задача в том, чтобы определить конкретный тип мутации, встречающейся в семье, и выявить всех носителей. ДНК-тесты не являются обычным тестом, но они могут помочь диагностировать гемоглобинопатию и выявить носителей.
- Мазок периферической крови (микроскопический дифференциальный подсчет лейкоцитов, считываемый по мазку периферической крови). Тест проводится путем формирования тонкого слоя крови на предметном стекле и окрашивания его специальными красителями. Образец крови, обработанный таким образом, затем оценивается лаборантом под микроскопом. Специалист определяет количество и тип белых и красных кровяных телец и тромбоцитов. Оценивает, являются ли они нормальными и зрелыми.
Анализ крови
При гемоглобинопатии эритроциты могут быть в следующих формах:
- Микроциты (меньше нормального).
- Гипохромные (более бледные, с пониженным гемоглобином).
- Разных размеров (анизоцитоз) и формы (пойкилоцитоз, например, серповидно-клеточные клетки).
- С ядром (в незрелых эритроцитах) или с включениями.
- С неравномерным распределением гемоглобина (клетки-мишени, которые под микроскопом выглядят как «бычий глаз»).
Наличие более высокого процента аномально выглядящих эритроцитов означает более высокую вероятность наличия заболевания.
С помощью тестов на гемоглобинопатию и их комбинаций можно диагностировать наиболее распространенные гемоглобинопатии. Эти тесты могут помочь выявить пациентов с сочетанием различных гемоглобинопатий (смешанные гетерозиготы).
Лечение гемоглобинопатии
В настоящее время гемоглобинопатии – неизлечимые заболевания. Но возможно устранять симптомы заболевания. Цель – облегчить боль и минимизировать возможные осложнения. Также существуют лекарства, повышающие уровень гемоглобина F, что облегчает некоторые симптомы.
Однако исследования и поиск более безопасных и эффективных методов лечения все еще продолжается. В будущем для восстановления мутированного гена можно будет использовать трансплантацию стволовых клеток или генную терапию. Для того чтобы эти методы могли широко использоваться в будущем, необходимы дальнейшие обширные исследования.
Источники: БЕРТИС, Калифорния, ЭШВУД, Эр., Брунс, Делавэр, (ред.), Учебник Тиц по клинической химии и молекулярной диагностике. 4-е издание Луи: Эльзевье-Сондерс, 2006; LOTHAR, T. Клиническая лабораторная диагностика. Франкфурт: TH-Books, 1998; MASOPUST, J. Клиническая биохимия – требования и оценка биохимических исследований, часть I. и часть 2, Прага: Каролинум, 1998; RACEK, J., et al. Клиническая биохимия. 2. переработанное издание, Прага: Гален, 2006; Каспер Д.Л., Браунвальд Э., Фаучи А.С., Хаузер С.Л., Лонго Д.Л., редакторы Джеймсон Д.Л., 2005.
Поделиться ссылкой:
Источник