Анемия связан с хромосомой

Наследственные сидеробластные анемии – варианты, диагностика, лечениеНаследственные сидеробластные анемии — разнородная по спектру генетических нарушений и выраженности клинических проявлений группа заболеваний. Варианты наследования и патогенезНаследственные сидеробластные анемии, сцепленные с Х-хромосомой. У большинства детей с сидеробластной анемией механизм наследования связан с Х-хромосомой. В связи с этим анемия диагностируется преимущественно у мальчиков и их близких родственников мужского пола по материнской линии (родные дяди и двоюродные братья). Редко имеется другой вариант наследования, не связанный с Х-хромосомой. В некоторых семьях заболевание возникает только у девочек, так как у мальчиков-гомозигот патология не совместима с жизнью. В основе патогенеза лежит дефект синтеза 5-аминолевулинатсинтетазы (АЛК), участвующей в синтезе гема. Для нормальной работы этого фермента необходимо достаточное количество пиридоксина — витамина В6 (этим и объясняется эффективность лечения витамином В6). Активность фермента 5-аминолевулинатсинтетазы (АЛК-синтетазы) кодирует ген ALAS2, расположенный на Х-хромосоме. У больных сидеробластной анемией описано несколько различных мутаций данного гена. Наследственные сидеробластные анемии с аутосомным типом наследования. Этот тип наследования встречается значительно реже, чем сцепленный с Х-хромосомой. Известны случаи как аутосомно-доминантного, так и аутосомно-рецессивного наследования. Мутации гена ALAS2 не определяются, поэтому терапевтический эффект пиридоксина отсутствует. Спорадическая врожденная сидеробластная анемия. В мире описано около 20 случаев сидеробластной анемии, выявленной сразу после рождения, без признаков заболевания у других членов семьи. Вероятно, при этом имелись либо аутосомно-рецессивный тип наследования, либо появление новых мутаций гена ALAS2 в родительских половых клетках. Митохондриальная цитопатия (синдром Пирсона). Синдром Пирсона — врожденное заболевание, которое обусловлено делециями или другими генетическими перестройками митохондриальной ДНК и клинически проявляется множественными органными поражениями. Одним из ранних признаков заболевания является тяжелая анемия, ассоциированная с наличием кольцевых сидеробластов в костном мозге. Продолжительность жизни детей с синдромом Пирсона обычно не превышает 2-3 года. Синдром DIOMOAD. В основе большинства клинических проявлений заболевания, наследующегося по аутосомно-рецессивному типу, лежат дегенеративные процессы в нервной ткани, обусловленные, вероятно, наследственными дефектами метаболизма тиамина. Гематологическим проявлением синдрома является нормоцитарная сидеробластная анемия средней степени тяжести в сочетании с нейтропенией и выраженной тромбоцитопенией. Отмечен терапевтический эффект применения тиамина. Классификация сидеробластных анемий Данные клинических и лабораторных исследований при наследственных сидеробластных анемийВ большинстве случаев клиническая картина сидеробластной анемии, сцепленной с Х-хромосомой, не отличается от таковой при анемии с аутосомным типом наследования или других врожденных форм. Тяжелая анемия, как правило, диагностируется в младенчестве или раннем детстве. При менее выраженных проявлениях анемического синдрома или бессимптомном течении заболевание выявляется обычно у взрослых или даже пожилых больных. Наряду с анемическим синдромом у всех больных определяются признаки избытка железа в организме, которые получили специальное название: синдром эритропоэтигеского гемохроматоза. Проявления этого синдрома достаточно разнообразны. Чаще всего определяются умеренная гепатомегалия и спленомегалия. Функция печени в большинстве случаев нарушена незначительно или не страдает вообще. При биопсии печеночной ткани определяются депозиты железа в гепатоцитах. У некоторых больных старше 30-40 лет при гистологическом исследовании находят признаки микронодулярного цирроза печени, протекающего, как правило, доброкачественно. На фоне гемохроматоза поджелудочной железы могут определяться сахарный диабет или нарушения толерантности к глюкозе. Редко при объективном обследовании выявляется пигментация кожных покровов. Наиболее опасными проявлениями синдрома эритро-поэтического гемохроматоза являются тяжелые нарушения сердечного ритма и сердечная недостаточность, которые развиваются, как правило, на поздних этапах заболевания. В тяжелых случаях у детей может наблюдаться задержка роста и развития. В анализе крови определяется анемия различной степени тяжести. При тяжелой анемии обычно имеются гипохромия, микроцитоз, анизо- и пойкилоцитоз, реже мишеневидные клетки и единичные сидероциты. При менее тяжелых формах анемии в мазке могут выявляться две популяции клеток: гипохромные микроциты и нормальные эритроциты. На гистограмме эритроцитов при этом образуется двухфазная кривая, отражающая различия в размерах эритроцитов. Уровень лейкоцитов и тромбоцитов обычно в норме, но при развитии гиперспленизма может снижаться. Количество ретикулоцитов в большинстве случаев в норме или незначительно повышено. В миелограмме выявляется гиперплазия эритроидного ростка на фоне нормобластического типа кроветворения и повышенное количество сидеробластов. В редких случаях (при сопутствующем дефиците фолиевой кислоты) может определяться мегалобластический тип кроветворения. При биохимическом исследовании обычно выявляется незначительная гипербилирубинемия, повышение уровня ферритина и снижение уровня трансферрина, и увеличение его сатурации. При анемии, сцепленной с Х-хромосомой, имеется снижение активности АЛК-синтетазы.
У больных с синдромом Пирсона диагностируется рефрактерная сидеробластная анемия, которая сочетается с признаками экзокринной недостаточности поджелудочной железы, эпизодами молочнокислого ацидоза и прогрессирующей почечной и печеночной недостаточностью. Анемия выявляется, как правило, после рождения и носит нормоцитарныи или макроцитарный характер. Уровень ретикулоцитов чаще всего снижен. В большинстве случаев имеются нейтропения и тромбоцитопения различной степени тяжести. При электрофорезе гемоглобина обычно определяется повышение уровня Hb F. Костный мозг гиперклеточный или нормоклеточный, выявляются кольцевые сидеробласты. Лечение и прогноз наследственных сидеробластных анемийВсем больным наследственными сидеробластными анемиями необходимо начинать терапию пиридоксином (витамин В6), которая эффективна в среднем в 1/3 случаев. Доза витамина В6 обычно составляет 50-100 мг в сутки. Выраженность терапевтического эффекта у пациентов, ответивших на лечение, различна. В большинстве случаев появляется ретикулоцитоз и уровень гемоглобина в течение 1-2 месяцев лечения постепенно повышается до нормы или субнормальных значений. Морфологические изменения эритроцитов при этом сохраняются. При отсутствии оптимального эффекта на фоне терапии пиридоксином уровень гемоглобина стабилизируется, но не достигает нормальных значений. Необходимо проведение поддерживающей терапии витамином В6, при отсутствие которой концентрация гемоглобина через несколько месяцев снижается до исходных величин. При выявлении мегалобластического типа кроветворения показано лечение фолиевой кислотой. Пациентам с тяжелой анемией, у которых отсутствует эффект витамина В6, по показаниям проводятся периодические трансфузии эритроцитарной массы. Это уменьшает выраженность анемии и предотвращает задержку роста и развития у детей. Для профилактики гемосидероза показана терапия дефероксамином, в которой нуждаются прежде всего больные с тяжелой анемией, получающие гемотрансфузионную терапию. Предпочтительно проведение 12-часовых подкожных инфузий препарата в дозе 40 мг/(кг-день) каждые 5 дней недели (этот режим обладает минимальной токсичностью). Спленэктомия при наследственных формах сидеробластной анемии довольно часто осложняется тромбоэмболией, в ряде случаев с летальным исходом, поэтому оперативное лечение используется редко. – Также рекомендуем “Идиопатическая приобретенная сидеробластная анемия – причины, диагностика, лечение” Оглавление темы “Анемии”:
|

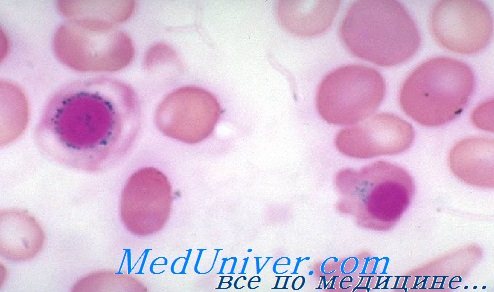
Наследственная сидеробластическая анемия – анемия, реагирующая на пиридоксинНаследственная сидеробластическая анемия описана Gooley и Rundles. Первые выявленные случаи четко указывали на отношение к мужскому полу, в связи с чем их назвали и «связанных с полом», причем передача этой анемии рецессивная. Преимущественное поражение мужчин подсказывает мысль о том, что нарушение наследственного характера, составляя рецессивную черту, связанную с хромосомой X. У женщин-носителей выявлены гипохромные эритроциты. В этих аномальных эритроцитах обнаруживалось также наличие связанного с хромосомой X антигена — Хга, в то время как нормальные эритроциты не содержали такого антигена. Результаты наблюдения рассматривались как способ передачи, связанный с хромосомой X (Cartwright, Lee). Такого рода анемии развиваются у подростков, им характерны признаки сидеробластической анемии с выраженной эритроцитной гипохромией, а весьма часто — и гемохроматозом. Гейльмейер, назвавший это заболевание «наследственной сидеробластической анемией», исследовал также метаболизм гема данных больных, обнаружив недостаток копропорфириноксидазы. По этой причине количество протопорфирина в эритроцитах меньше, в то время как копропорфирина — в избытке.
Отдельными авторами был отмечен и недостаток дельтааминолевулинсинтетазы. Эти биохимические аномалии были обнаружены и у членов соответствующих семей. Гейльмейер описал также вид наследственной болезни, не связанной с полом (II тип), в которой нарушение происходит на первой фазе синтезирования протопорфирина, именно при переходе а-амино-b-кетоадипиновой кислоты в дельта-аминолевулиновую кислоту. В моче обнаружен эстер (гидразон) а-амино-b-кетоадипиновой кислоты. Лечение заключается в назначении крупных доз (250 мг/сутки) пиридоксина, фолиевой кислоты, комплексонов железа (десферал), а в случае надобности и переливания крови. Анемия, реагирующая на пиридоксинАнемия, реагирующая на пиридоксин – этим названием обозначена крупная группа анемий, которые, морфологически, похожи на наследственные анемии (эритроцитная гипохромия, кольчатые сидеробласты, гемохроматоз и пр.), «поддающиеся лечению крупными дозами пиридоксина». Этот вид анемии не развивается за счет недостатка пиридоксина, уровень которого в крови нормальный, но видимо он не преобразуется в активную форму — пиридоксалфосфат (Beaupre и Growney). Чрезмерная нагрузка триптофаном обусловливает появление в моче промежуточных продуктов его катаболизма (за отсутствием активного пиридоксина), таких как кинуренин и ксантуреновая кислота. Внимательный анализ ряда случаев этой анемии выявил бесспорно семейный характер заболевания (Medal и сотр.). В настоящее время проявляется тенденция включения этой болезни в крупную группу наследственных сидеробластических анемий, которые, как было доказано, в размере 50% дают положительный ответ на лечение пиридоксином крупными дозами (т.е. примерно 250—300 мг/сутки, в то время как 5 мг/сутки пиридоксина в принципе назначаются для приведения к норме недостатка пиридоксина, например при отравлении ГИНК и пр.). – Также рекомендуем “Вторичная сидеробластическая анемия – анемии, вызываемые изониазидотерапией и отравлением свинцом” Оглавление темы “Гематология”:
|
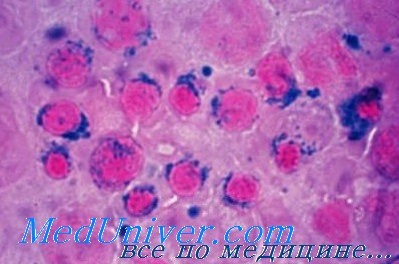

Анемия Фанкони (АФ), или панцитопения Фанкони, является синдромом нестабильности генома. Это редкое наследственное аутосомно-рецессивное заболевание с вариабельной пенетрантностью и генетической гетерогенностью. АФ была впервые описана в 1927 г. швейцарским педиатром Гвидо Фанкони, который сообщил о 3 братьях с панцитопенией и пороками физического развития. Термин «анемия Фанкони» был предложен Негели в 1931 г. для обозначения комбинации семейной анемии Фанкони и врожденных физических пороков. По настоящее время описано чуть более 2000 случаев АФ. Частота гетерозиготного носительства существенно различается в разных популяциях. Традиционно указывалась цифра 1:300, по последним данным североамериканского регистра, она составляет 1:181, в Израиле — 1:93. Следует заметить, что около 6 % больных не имеют никаких аномалий развития. Некоторые мутации этнически закреплены, в основе их распространения лежит «эффект основателя» — потеря генетической вариабельности в популяциях, основанных малым количеством предков, что свойственно для относительно небольших популяций. Эти мутации встречаются у евреев-ашкенази, испанских цыган, голландцев, выходцев с Канарских островов, у жителей Южной Африки и Кореи. В некоторых из этих популяций частота носительства этнически закрепленных мутаций довольно высока и оценена приблизительно как 1:100. Частота встречаемости тех или иных мутаций в РФ не изучена.
Этиология и патогенез
Нарушение структуры ДНК является результатом воздействия как внутренних (депуринизация, дезаминирование, воздействие эндогенных альдегидов, особенно формальдегида, и активных форм кислорода), так и внешних факторов (ионизирующее и УФ облучение, химические мутагены). При воздействии этих факторов происходят реакции алкилирования, окисления, восстановления, связывания с формальдегидными группами азотистых оснований. В итоге возникают изменения одного или нескольких оснований, вставки и делеции, образование тиминовых димеров, одно- и двухцепочечные разрывы ДНК, образование сшивок между основаниями одной цепи или комплементарными цепями ДНК, между ДНК и белковыми молекулами. Несмотря на это, в целом геном остается свободным от «ошибок», так как клетка имеет механизмы детекции и репарации поврежденной ДНК. Поврежденное основание может быть восстановлено непосредственно его заменой или обратной химической реакцией (direct repair), в других случаях необходимы более сложные процессы, обеспечивающие удаление поврежденного участка ДНК и достраивание правильной последовательности с использованием комплементарной цепи, редко — гомологичной хромосомы. Такая репарация ДНК — сложный многоступенчатый процесс взаимодействия нескольких каскадных путей. Процесс репарации происходит на разных этапах клеточного цикла. При АФ нарушается способность клетки исправлять определенный тип повреждений ДНК — поперечные межхроматидные сшивки (DNA interstrand crosslink), которые препятствуют работе репликационной вилки. Поперечные межхроматидные сшивки формируются как под воздействием продуктов естественного метаболизма клетки (в первую очередь эндогенных альдегидов, но также и активных форм кислорода), так и под воздействием химических веществ, в частности химиотерапевтических препаратов. К такому типу химических веществ относятся алкилирующие соединения, имеющие в своем составе две активные алкильные группы, обеспечивающие им активное связывание с определенными основаниями: цисплатин, митомицин С, азотистый иприт, псорален, диэпоксибутан, мелфалан, циклофосфамид, мустарген, стрептозоцин. Протеины, функция которых нарушается при АФ, задействованы во всех этапах репарации межхроматидной поперечной сшивки. Этот сложный многоступенчатый процесс получил название FA-pathway, а протеины, задействованные в нем, — АФ-протеины. Ключевую роль в этом процессе играет моноубиквитинирование гена FANCD2, который координирует процессы вырезки поврежденных нуклеотидов, прямое достраивание поврежденного участка и гомологичную рекомбинацию. При АФ клетка не способна адекватно исправлять повреждения ДНК, накопление поломок которой может приводить к костномозговой недостаточности, аномалиям физического развития и предрасположенности к развитию опухолей.
Спектр мутаций, которые приводят к АФ:
1) FANCA.
Мутации в этом гене — самые распространенные и встречаются в 60–70 % случаев АФ. Известно более 100 мутаций, из которых около трети приходится на точечные, еще треть представляют собой микроделеции, и около 40 % представлены крупными делециями. Описаны также и малые дупликации. По действию мутации в гене FANCA могут быть гипоморфными, т. е. приводить к частичной потере функции белка; они характеризуются более мягкими клиническими проявлениями, однако большая часть мутаций вызывает полную потерю функции. Ряд мутаций в гене FANCA имеет повышенную частоту распространения. Так, микроделеция в 38-м экзоне с.3788_3790delTCT — самая распространенная мутация при АФ в мире (20,7 % всех аллелей с мутацией). При этом она встречается у 80 % пациентов с АФ с Канарских островов, где частота встречаемости АФ достигает 1:16000 новорожденных. Кроме того, эта мутация встречается в 51 % случаев АФ в Бразилии. Для подтверждения «эффекта основателя» для данной мутации был проведен анализ гаплотипа пациентов путем изучения вариабельных тандемных повторов и однонуклеотидных полиморфизмов гена FANCA у 28 пациентов с мутацией с.3788_3790delTCT из различных частей света. Все, за исключением одного пациента, имели общего предка. По всей видимости, Канарские острова послужили местом происхождения и распространения заболевания из Европы в Америку, так как несколько веков назад практически все суда из Испании в Америку шли через Канарские острова. Однако, учитывая, что эта мутация тем не менее составляет 2–5 % всех FANCA-мутаций, существует также и мнение, что она связана с явлением существования определенных участков генома с повышенной мутационной способностью, так называемых hotspot. Другой пример «эффекта основателя» — мутация с.295С>Т, которую выявляют почти во всех случаях АФ у испанских цыган. При этом носительство этой мутации среди испанских цыган определено как 1:67. Функционально и фенотипически варианты мутаций в гене FANCA проявляются одинаково.
2) FANCC.
Мутации в гене FANCC встречаются в 10–15 % случаев АФ, почти 90 % случаев представлено двумя мутациями — с.711+4А>Т и delG332. Самая частая мутация в гене FANCC — с. 711+4А>Т, ее выявляют в гомозиготном состоянии в 80 % случаев АФ у евреев-ашкенази. Частота гетерозиготного носительства этой мутации среди евреев-ашкенази достигает 40 %. При этом встречаются и спорадические случаи. Генотип FANCC с.711+4А>Т также распространен среди больных АФ в Японии. В Нидерландах более чем 50 % случаев АФ — это гомозиготные носители мутации с.67delG (также известна как delG332), приводящей к сдвигу рамки считывания в гене FANCC.
3) FANCG.
FANCG задействован в 10 % случаев АФ. Встречаются практически все типы мутаций, за исключением крупных делеций. Клинически характеризуются более частым и быстрым развитием миелодисплазии или лейкоза. Встречаются как спорадические случаи, так и этнически ассоциированный вариант — около 80 % случаев АФ у чернокожих южноафриканцев (Bantu-speakers) (Южная Африка, Свазиленд, Малави и Мозамбик) имеют мутацию 637_643delTACCGCC. Заболевание характеризуется частыми нарушениями пигментации кожи, слабовыраженными аномалиями развития и сравнительно поздними, но тяжелыми гематологическими проявлениями, обусловленными в том числе и поздним обращением к врачу.
Развитие костномозговой недостаточности связывают с повышенным апоптозом гемопоэтических клеток, однако истинные патогенетические механизмы костномозговой недостаточности при АФ мало изучены из-за сложности получения адекватной биологической модели развития заболевания. Последние исследования на ксенографтных моделях и in vitro показали, что в ответ на накопление нерепарированных повреждений ДНК происходит активация р53 проапоптотического пути и запуск поздней р21(Cdkn1a)-зависимой блокировки клеточного цикла в фазе G0/G1 с последующей элиминацией ранних гемопоэтических предшественников из костного мозга. Этот механизм запускается в пренатальном периоде на этапе формирования пула стволовых клеток и ранних клеток-предшественников гемопоэза, что приводит к значительному снижению их количества. Накопление дефектов ДНК после рождения в результате различных физико-химических воздействий усугубляет нарушение гемопоэза. Кроме выраженного апоптоза ранних клеток-предшественников происходит нарушение базовых свойств стволовых кроветворных клеток — способности к самоподдержанию собственной популяции, пролиферации и дифференцировке в различные линии гемопоэза. Генетическая нестабильность при АФ реализуется в повышенной частоте развития ряда опухолей, наиболее частые — ОМЛ и плоскоклеточный рак кожи головы и шеи, слизистых оболочек рта и мочеполового тракта — то есть тканей, характеризующихся высокой пролиферативной активностью.
Клиническая картина
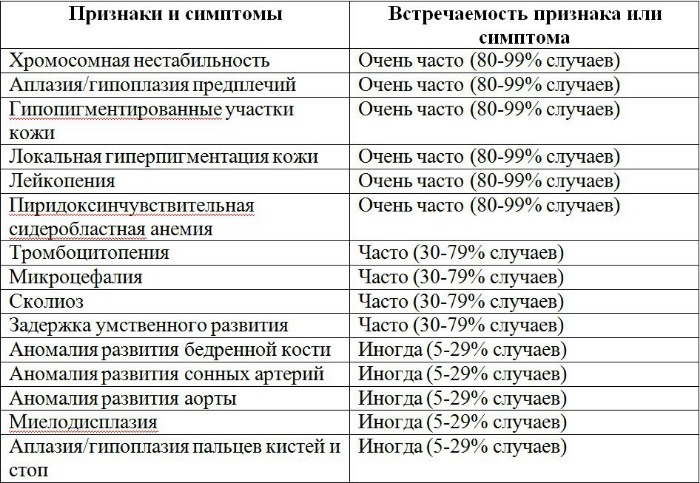
Признаки и симптомы, а также частота их встречаемости указаны в таблице 1.
Таблица 1 | Признаки и симптомы анемии Фанкони
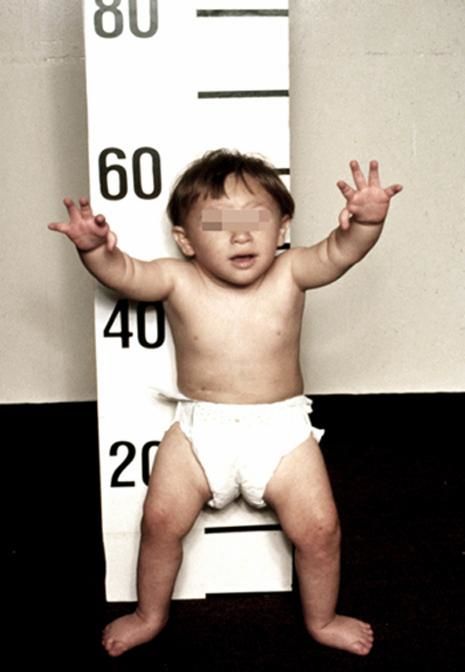
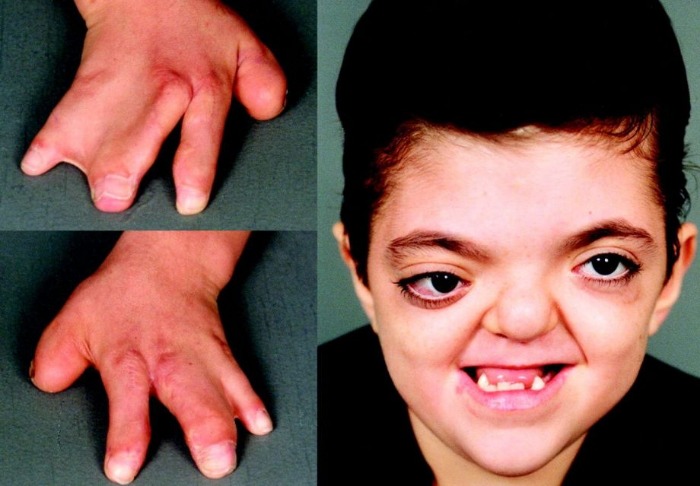
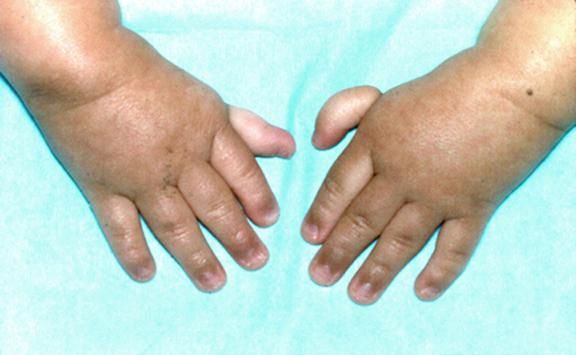
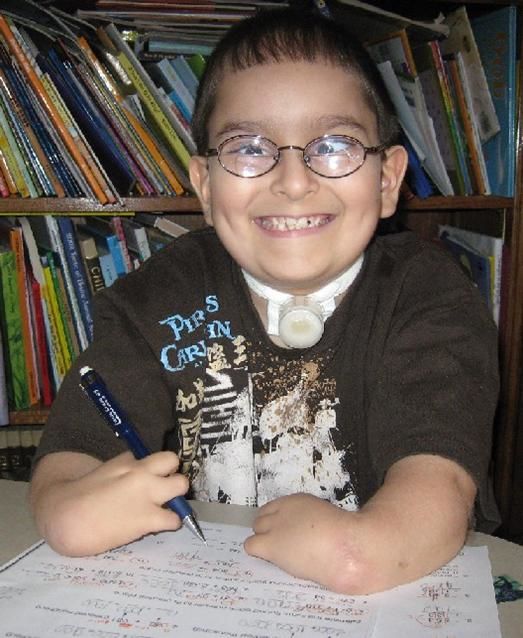
Рисунки 1–5 | Пороки развития, наблюдаемые при анемии Фанкони.
Диагностика
Помимо тщательного физикального осмотра, сбора семейного анамнеза, общего анализа крови и пункции костного мозга для более точной диагностики используются такие методы лабораторной диагностики, как тест на ломкость хромосом, метод MLPA, высокопроизводительное секвенирование и секвенирование по Сэнгеру.
1) Тест на ломкость хромосом.
«Золотым стандартом» скрининга для выявления АФ был и остается тест с диэпоксибутаном (1,3-butadienediepoxide) и его вариант с митомицином С (MMC). Еще в начале изучения АФ было отмечено, что фибробласты и лимфоциты больных АФ в культуре клеток демонстрируют спонтанную повышенную ломкость хромосом. Значительные различия в уровне спонтанных аберраций у больных (вплоть до отсутствия таковых) требовали унифицированного и точного метода детекции. Позже была показана повышенная чувствительность клеток больных АФ к действию алкилирующих агентов, вызывающих поперечные сшивки между нуклеотидами, что препятствует образованию нормальной репликативной вилки для запуска процесса репарации ДНК, позже получивших общее название interstrand cross-link agent. На основании этого был предложен цитогенетический метод диагностики АФ: после обработки лимфоцитов или фибробластов алкилирующим веществом (в нелетальной для клеток концентрации) определяют частоту и спектр спонтанных и индуцированных in vitro хромосомных аберраций. Обычно ставится несколько параллельных клеточных культур, стимулированных фитогемагглютинином лимфоцитов периферической крови: без добавления алкилирующего агента (для определения спонтанного уровня аберраций) и с его добавлением в разной концентрации. Затем в метафазных пластинках подсчитывают число хромосомных разрывов. Для АФ характерны разрывы хромосом с образованием радиальных фигур, фрагментов, хромосомных и хроматидных разрывов, а также три- и тетрарадикалов. При анализе результатов учитывают число разрывов по отношению к числу проанализированных метафаз, процент клеток с разрывами и ряд других показателей. Необходимо отметить, что значения, при которых тест на ломкость хромосом считается положительным, в различных лабораториях варьируются. Тест не имеет 100 % специфичности. Положительный тест на ломкость хромосом бывает у пациентов с синдромом Ниймеген (мутации в гене NBS1), синдромом Робертса (мутации в гене ESCO2), Warsaw breakage syndrome (мутации в гене DDX11), синдромом Блюма (ген BLM), врожденном дискератозе и некоторых других синдромах. В то же время АФ-подобные синдромы могут оказаться действительно АФ с соответствующим генетическим дефектом.
2) Секвенирование по Сэнгеру долгие годы было основным методом определения мутаций при АФ. Учитывая значительные размеры генов, секвенирование каждого из них представляет собой довольно трудоемкий и дорогостоящий процесс. Его обычно проводили после анализа на группу комплементации, предварительно определив вероятный ген. Анализ нуклеотидной последовательности для всех известных АФ-генов затруднен количеством возможных мутаций в каждом, их разнообразием, в том числе в виде крупных инсерций или делеций (indel-мутации). Их длина может варьировать от одного до нескольких сотен и даже тысяч нуклеотидных оснований, что подразумевает использование совершенно разных методов молекулярно-генетического исследования.
3) Метод MLPA (мультиплексная амплификация лигазносвязанных проб) предназначен для определения делеций и амплификаций определенных последовательностей гена длиной до нескольких десятков нуклеотидов. Одновременно может быть исследовано до 60 таких последовательностей, что позволяет выявить как сравнительно небольшие делеции, так и делеции отдельных экзонов и целого гена. Метод MLPA используют для инициального скрининга делеции в гене FANCA, параллельно ген FANCA полностью секвенируют. Если пациент мужского пола, его исследуют на наличие делеций ген FANCB. Выявленные делеции желательно подтвердить другим методом. При этом методы, которые могут быть использованы, требуют индивидуальной разработки в каждом конкретном случае: количественная ПЦР, ПЦР длинных фрагментов (long range PCR) и хромосомный микроматричный анализ.
4) Высокопроизводительное секвенирование.
Метод, который позволяет одномоментно анализировать от нескольких генов до полного генома, является наиболее подходящим для определения мутаций при АФ. Возможны несколько вариантов теста. Первый — секвенирование экзома, позволяет получить максимальный объем информации. Второй — секвенирование ограниченного числа интересующих и уже описанных в литературе генов (таргетное ресеквенирование), список которых можно дополнять или моделировать в соответствии с потребностями исследования. Современные коммерческие панели генов, как правило, помимо генов АФ включают большое число генов, ответственных за развитие других врожденных синдромов, в том числе и АФ-подобных. Внедрение в практику высокопроизводительного секвенирования позволяет избежать последовательного трудоемкого исследования каждого из известных генов методом секвенирования по Сэнгеру, однако пока не позволяет с должной уверенностью выявить крупные делеции и дупликации. Найденные мутации требуют подтверждения одним из других подходящих методов. При обнаружении новых мутаций необходимо подтверждение их патогенности в функциональном тесте и др.
Пренатальная и преимплантационная диагностика должна проводиться в первую очередь в семьях, где ранее были установлены патогенетические мутации. В этом случае проводится целенаправленный поиск известной мутации. Материалом для диагностики служат клетки плода, получаемые путем биопсии ворсин хориона на 10–12 неделе беременности. Следует помнить, что генетический анализ занимает не менее 2–3 недель. Если нет возможности провести молекулярно-генетическое исследование, возможно выполнение теста на ломкость хромосом клеток ворсин хориона на 10–12 неделе беременности либо при амниоцентезе на 15–18 неделе. Однако молекулярно-генетическое исследование предпочтительнее.
Лечение
Основной метод лечения АФ — аллогенная трансплантация гемопоэтических стволовых клеток.
Из медикаментозного лечения в настоящий момент применяют андрогены. Препараты этой группы (оксиметалон, метандростенолон) позволяют достичь гематологического ответа примерно у 50 % больных. Также применение андрогенов значимо увеличивает продолжительность жизни у ответивших на лечение пациентов: медиана продолжительности жизни составляет 9 лет после установления диагноза против 2,5 лет соответственно для тех пациентов, у которых лечение андрогенами не было эффективным.
При возникновении сопутствующих заболеваний, например, инфекций, обосновано применение гранулоцитарного колониестимулирующего фактора, который способен временно увеличить количество нейтрофилов и облегчить протекание заболевания.
В 2015 году коллективу ученых удалось успешно применить технологию Crispr/Cas9 для редактирования мутации с.67delG гена FANCC.
Источники:
- https://www.bloodjournal.org/content/126/23/3622?sso-checked=true
- https://www.ncbi.nlm.nih.gov/books/NBK1401/
- https://rarediseases.info.nih.gov/diseases/6425/fanconi-anemia
- https://rarediseases.org/rare-diseases/fanconi-anemia/
- https://cyberleninka.ru/article/v/geneticheskaya-diagnostika-anemii-fankoni-obzor-literatury