Что такое редуцированный гемоглобин

Оглавление темы “Функции клеток крови. Эритроциты. Нейтрофилы. Базофилы.”: Гемоглобин. Типы ( виды ) гемоглобина. Синтез гемоглобина. Функция гемоглобина. Строение гемоглобина.Гемоглобин — это гемопротеин, с молекулярной массой около 60 тыс., окрашивающий эритроцит в красный цвет после связывания молекулы O2 с ионом железа (Fe++). У мужчин в 1 л крови содержится 157 (140—175) г гемоглобина, у женщин — 138 (123—153) г. Молекула гемоглобина состоит из четырех субъединиц гема, связанных с белковой частью молекулы — глобином, сформированной из полипептидных цепей.
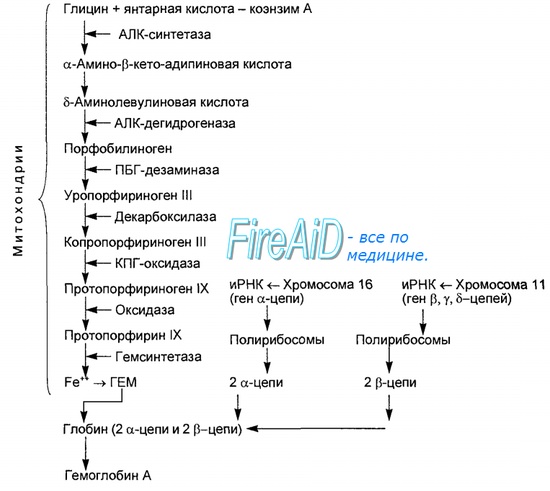
Синтез гема протекает в митохондриях эритробластов. Синтез цепей глобина осуществляется на полирибосомах и контролируется генами 11-й и 16-й хромосом. Схема синтеза гемоглобина у человека представлена на рис. 7.2. Гемоглобин, содержащий две а- и две В-цепи, называется А-тип (от adult — взрослый). 1 г гемоглобина А-типа связывает 1,34 мл O2. В первые три месяца жизни плода человека в крови содержатся эмбриональные гемоглобины типа Gower I (4 эпсилон цепи) и Gower II (2а и 25 цепи). Затем формируется гемоглобин F (от faetus — плод). Его глобин представлен двумя цепями а и двумя В. Гемоглобин F обладает на 20—30 % большим сродством к O2, чем гемоглобин А, что способствует лучшему снабжению плода кислородом. При рождении ребенка до 50—80 % гемоглобина у него представлены гемоглобином F и 15—40 % — типом А, а к 3 годам уровень гемоглобина F снижается до 2 %. Соединение гемоглобина с молекулой 02 называется оксигемоглобином. Сродство гемоглобина к кислороду и диссоциация оксигемоглобина (отсоединения молекул кислорода от оксигемоглобина) зависят от напряжения кислорода (Р02), углекислого газа (РС02) в крови, рН крови, ее температуры и концентрации 2,3-ДФГ в эритроцитах. Так, сродство повышают увеличение Р02 или снижение РС02 в крови, нарушение образования 2,3-ДФГ в эритроцитах. Напротив, повышение концентрации 2,3-ДФГ, снижение Р02 крови, сдвиг рН в кислую сторону, повышение РС02 и температуры крови — уменьшают сродство гемоглобина к кислороду, тем самым облегчая ее отдачу тканям. 2,3-ДФГ связывается с р-цепями гемоглобина, облегчая отсоединение 02 от молекулы гемоглобина. Увеличение концентрации 2,3-ДФГ наблюдается у людей, тренированных к длительной физической работе, адаптированных к длительному пребыванию в горах. Оксигемоглобин, отдавший кислород, называется восстановленным, или дезоксигемоглобином. В состоянии физиологического покоя у человека гемоглобин в артериальной крови на 97 % насыщен кислородом, в венозной — на 70 %. Чем выраженней потребление кислорода тканями, тем ниже насыщение венозной крови кислородом. Например, при интенсивной физической работе потребление кислорода мышечной тканью увеличивается в несколько десятков раз и насыщение кислородом оттекающей от мышц венозной крови снижается до 15 %. Содержание гемоглобина в отдельном эритроците составляет 27,5—33,2 пикограмма. Снижение этой величины свидетельствует о гипохромном (т. е. пониженном), увеличение — о гиперхромном (т. е. повышенном) содержании гемоглобина в эритроцитах. Этот показатель имеет диагностическое значение. Например, гиперхромия эритроцитов характерна для В|2-дефицитной анемии, гипохромия — для железодефицитной анемии. – Также рекомендуем “Старение эритроцитов. Разрушение эритроцитов. Длительность жизни эритроцита. Эхиноцит. Эхиноциты.” |
Виды гемоглобина.
Различают три вида гемоглобина; первоначально эмбрион имеет примитивный гемоглобин (HbP) – до 4-5 мес. внутриутробной жизни, затем начинает появляться фетальный гемоглобин (HbF), количество которого увеличивается до 6–7 мес. внутриутробной жизни. С этого срока происходит увеличение гемоглобина А (взрослого) максимальная величина которого достигает к 9 мес. внутриутробной жизни (90%). Количество фетального гемоглобина при рождении является одним из признаков доношенности: чем больше HbF, тем менее доношенный ребенок. Следует отметить, что HbF в присутствии 2,3 дифосфоглицерата (ДФГ – продукт метаболизма оболочки эритроцита при недостатки кислорода) не меняет своего сродства к кислороду в отличии от HbA, сродство которого к кислороду снижается.
Виды Нв отличаются друг от друга по степени химического сродства к О2. Так, НвF в физиологических условиях имеет более высокое сродство к О2, чем НвА. Эта важнейшая особенность НвF создает оптимальные условия для транспорта О2 кровью плода.
Гемоглобин представляет собой кровяной пигмент, роль которого заключается в транспорте кислорода к органам и тканям, транспорте двуокиси углерода от тканей к легким, кроме этого он является внутриклеточным буфером, который поддерживает оптимальную для метаболизма pH. Гемоглобин содержится в эритроцитах и составляет 90% их сухой массы. Вне эритроцитов гемоглобин практически не обнаруживается.
Химически гемоглобин относится к группе хромопротеидов. Его простетическая группа, включающая железо, называется гемом, белковый компонент – глобином. Молекула гемоглобина содержит 4 гема и 1 глобин.
К физиологическим гемоглобинам относятся НЬА (гемоглобин взрослого) и HbF (фетальный гемоглобин, составляющий основную массу гемоглобина плода и исчезающий почти полностью ко 2-му году жизни ребенка). Современными электрофоретическими исследованиями доказано существование по крайней мере двух разновидностей нормального гемоглобина А: А1 (главный) и А2 (медленный). Основную массу гемоглобина взрослого (96-99%) составляет HbAl, содержание других фракций (А2 F) не превышает 1 – 4%. Каждый вид гемоглобина, вернее его глобиновая часть, характеризуется своей «полипептидной формулой». Так, HbAl обозначается как ά2 β2, то есть он состоит из двух ά-цепей и двух β-цепей (всего 574 аминокислотных остатка, расположенных в строго определенном порядке). Другие виды нормальных гемоглобинов – F, A2 обладают общей с HbAl β-пептидной цепью, но отличаются структурой второй полипептидной цепи (например, структурная формула HbF – ά2γ2).
Помимо физиологических гемоглобинов, существуют еще несколько патологических разновидностей гемоглобина. Патологические гемоглобины возникают в результате врожденного, передаваемого по наследству дефекта образования гемоглобина.
В эритроцитах циркулирующей крови гемоглобин находится в состоянии беспрерывной обратимой реакции. Он то
присоединяет молекулу кислорода (в легочных капиллярах), то отдает ее (в тканевых капиллярах).
К основным соединениям гемоглобина относятся: ННв – восстановленный гемоглобин и НвСО2 – соединение с углекислым газом (карбогемоглобин). Они в основном находятся в венозной крови и придают ей темно-вишневый цвет.
НвО2 – оксигемоглобин– находится, в основном, в артериальной крови, придавая ей алый цвет. НвО2 – чрезвычайно нестойкое соединение, его концентрация определяется парциальным давлением О2 (рО2): чем больше рО2, тем больше образуется НвО2 и наоборот. Все вышеперечисленные соединения гемоглобина относятся к физиологическим.
Гемоглобин в венозной крови с низким парциальным давлением кислорода связан с 1 молекулой воды. Такой гемоглобин называется редуцированным (восстановленным) гемоглобином. В артериальной крови с высоким парциальным давлением кислорода гемоглобин соединен с 1 молекулой кислорода и имеет название – оксигемоглобин. Путем непрерывного превращения оксигемоглобина в редуцированный гемоглобин и обратно осуществляется перенос кислорода из легких к тканям. Восприятие углекислоты в тканевых капиллярах и доставка ее в легкие также является функцией гемоглобина. В тканях оксигемоглобин, отдавая кислород, превращается в редуцированный гемоглобин. Кислотные свойства редуцированного гемоглобина в 70 раз слабее свойств оксигемоглобина, поэтому свободные валентности его связывают углекислоту. Таким образом, углекислота доставляется из тканей в легкие с помощью гемоглобина. В легких образующийся оксигемоглобин в силу своих высоких кислотных свойств вступает в связь с щелочными валентностями карбогемоглобина, вытесняя углекислоту. Так как основной функцией гемоглобина является обеспечение тканей кислородом, то при всех состояниях, сопровождающихся снижением концентрации гемоглобина в крови, или при качественных его изменениях развивается гипоксия тканей.
Однако есть и патологические формы гемоглобина.
Гемоглобин обладает способностью вступать в диссоциирующие соединения не только с кислородом и углекислый газом, но и с другими газами. В результате образуются карбоксигемоглобин, оксиазотистый гемоглобинисульфгемоглобин.
Карбоксигемоглобин (оксиуглеродный) диссоциирует в несколько сотен раз медленнее, чем оксигемоглобин, поэтому даже незначительная концентрация (0,07%) в воздухе угарного газа (СО), связывая около 50% имеющегося в организме гемоглобина и лишая его способности переносить кислород, является смертельным. Карбоксигемоглобин (НвСО) – очень прочное соединение с угарным газом, обусловленное химическими свойствами угарного газа по отношению к Нв. Оказалось, что его родство к Нв в 400-500 раз больше, чем сродство О2 к Нв. Поэтому при незначительном повышении концентрации СО в окружающей среде образуется очень большое количество НвСО. Если в организме находится много НвСО, то возникает кислородное голодание. Фактически О2 в крови очень много, а клетки тканей его не получают, т.к. НвСО – прочное соединение с О2.
Метгемоглобин представляет собой более стойкое, чем оксигемоглобин, соединение гемоглобина с кислородом, получающееся при отравлениях некоторыми лекарственными препаратами – фенацетином, антипирином, сульфаниламидами. При этом двухвалентное железо простетической группы, окисляясь, превращается в трехвалентное. Метгемоглобин (MetНв) – окисленная форма Нв, крови придает коричневую окраску. Образуется MetНв при действии на Нв любым окислителями: нитраты, перекиси, перманганат калия, красная кровяная соль и т.д. Это стойкое соединение, потому что железо из ферроформы (Fe++) переходит в ферриформу (Fe+++), необратимо связывающую О2. При образовании в организме больших количеств MetНв также возникает кислородная недостаточность (гипоксия).
Сульфгемоглобин обнаруживается в крови иногда при применении лекарственных веществ (сульфаниламидов). Содержание сульфгемоглобина редко превышает 10%. Сульфгемоглобинемия – необратимый процесс. Так как пораженные эритроциты
разрушаются в те же сроки, что и нормальные, явлений гемолиза не наблюдается и сульфгемоглобин может находиться в крови в течение нескольких месяцев. На этом свойстве сульфгемоглобина основан метод определения сроков пребывания нормальных эритроцитов в периферической крови.
Дата добавления: 2015-11-05; просмотров: 4759 | Нарушение авторских прав | Изречения для студентов
Читайте также:
Рекомендуемый контект:
Поиск на сайте:
© 2015-2020 lektsii.org – Контакты – Последнее добавление
Гемоглобинопатия – это наследственные заболевания с единой проблемой – образованием аномальной формы гемоглобина, например, серповидноклеточная анемия S и талассемия.
Гемоглобинопатии носят эндемический характер – они возникают в определенном географическом районе, например, в Средиземноморье, Африке, Юго–Восточной Азии. В нашей стране они тоже встречаются.
Что такое гемоглобинопатия
Гемоглобинопатии – это заболевания, вызванные выработкой и присутствием аномальной формы гемоглобина.
Гемоглобин состоит из гема (частей, содержащих железо) и глобина (частей белка, состоящих из аминокислотных цепей). Молекулы гемоглобина (Hb или Hgb) находятся в красных кровяных тельцах. Их задача – связывать кислород в легких и передавать его тканям и органам, где они его выделяют.
Строение гемоглобина
Существует несколько типов цепей глобина: альфа, бета, дельта и гамма.
Типы нормального гемоглобина:
- A – HbA: составляет около 95-98% от общего гемоглобина у взрослых людей. Он содержит 2 альфа (α) цепи и две бета (β) цепи.
- A2 – HbA2: составляет около 2-3% от общего гемоглобина. Он содержит 2 цепи альфа (α) и две цепи дельта (δ).
- F (HbF): составляет около 2% от общего гемоглобина взрослого человека. Он содержит 2 альфа (α) цепи и две гамма (γ) цепи. Этот гемоглобин в основном вырабатывается у плода, его производство значительно снижается вскоре после рождения и достигает уровня взрослого человека в течение 1-2 лет.
К гемоглобинопатиям относятся: структурные варианты гемоглобина, гемоглобин S, серповидноклеточная анемия, гемоглобинопатия C, гемоглобинопатия E, талассемия, гемоглобин Бартс, наследственная персистенция гемоглобина плода.
Причины развития гемоглобинопатии
Гемоглобинопатии возникают в случае генетических изменений генов глобина, которые приводят к изменению аминокислот, составляющих белок глобина. Эти изменения влияют на:
- структуру гемоглобина, например, гемоглобин S, который вызывает серповидно-клеточную анемию;
- его поведение;
- количество продуцируемого вещества (талассемия);
- стабильность.
Серповидно-клеточная анемия
Существует четыре гена, кодирующих цепь альфа-глобина, и два гена, кодирующих цепь бета-глобина. Наиболее частым заболеванием, связанным с изменением альфа-цепи, является альфа-талассемия. Его тяжесть зависит от количества пораженных генов.
Талассемия характеризуется снижением продукции одной из цепей глобина, дисбалансом между альфа- и бета-цепями в гемоглобине A (альфа-талассемия) или увеличением малых форм, таких как Hb A2 или Hb F (бета-талассемия).
Изменения бета-цепей гемоглобина являются врожденными, аутосомно-рецессивными. Это означает, что больной человек должен иметь две дефектные копии гена, каждая от одного из родителей. Если один ген нормален, а другой дефектен, человек гетерозиготен, и мы называем его носителем. Аномальный ген может быть передан любому из потомков. Если рассматриваемый человек является гетерозиготным носителем, он может не иметь никаких симптомов и носительство не влияет на его здоровье.
Если происходят две модификации одного и того же бета-гена, человек гомозиготен по этому гену. Его организм может производить дефектный гемоглобин – возникает гемоглобинопатия с симптомами и потенциальными осложнениями. Степень тяжести зависит от генетической мутации и варьируется от случая к случаю. Копию гена можно передать потомству.
Если два аномальных бета-гена являются врожденными, человек является двойным, смешанным гетерозиготным. У него будут симптомы одной или обеих гемоглобинопатий. Один из аномальных бета-генов будет передаваться каждому из потомков.
Были идентифицированы сотни гемоглобинопатий в бета-цепях. Хотя лишь некоторые из них являются общими и клинически значимыми.
Клинические признаки и симптомы
Признаки и симптомы различаются по типу гемоглобинопатии и возможному сочетанию нескольких гемоглобинопатий. Некоторые приводят к усилению распада эритроцитов (гемолизу), уменьшению их общего количества и развитию анемии.
Клинические признаки включают:
- слабость, утомляемость;
- недостаток энергии;
- желтуха;
- бледность кожи.
Утомляемость
К серьезным клиническим признакам относятся:
- приступы сильной боли;
- удушье;
- увеличение селезенки;
- нарушения роста у детей;
- боль в верхней части живота (вызванная желчными камнями).
Удушье
Общие гемоглобинопатии
Красные кровяные тельца, содержащие аномальный гемоглобин, могут не переносить кислород достаточно эффективно. Они могут разрушаться раньше (чем в здоровых клетках крови) и развиваться гемолитическая анемия. Выявлены сотни гемоглобинопатий, но лишь некоторые из них являются общими и клинически значимыми.
Одной из наиболее распространенных гемоглобинопатий является серповидно-клеточная анемия с присутствием гемоглобина S. Это приводит к изменению формы – серповидно-клеточной – эритроцитов и снижению их выживаемости. Гемоглобин С может вызвать легкую гемолитическую анемию. Гемоглобин E обычно не приводит к развитию каких-либо или только очень легких клинических симптомов.
- Талассемия: самая распространенная генетическая аномалия в мире. Она часто встречается в Средиземноморье, на Ближнем Востоке и в Юго-Восточной Азии. Более легкая форма талассемии также встречается, например, у людей, родившихся в Чехии.
- Гемоглобин S: это основной гемоглобин людей с серповидно-клеточной анемией. В среднем эта мутация есть в одном из двух бета-генов у 8% американцев и африканцев. Возникновение этих мутаций в наших широтах встречаеся редко. Пациенты с заболеванием HbS имеют две аномальные цепи бета (b s) и две нормальные цепи альфа (a). Когда эритроциты, содержащие гемоглобин S, подвергаются действию пониженного количества кислорода (как это может быть в случае повышенной физической нагрузки или инфекционного заболевания легких), они деформируются, принимая форму полумесяца. Серповидные эритроциты могут блокировать периферические кровеносные сосуды и вызывать нарушения кровотока и боль. У них пониженная способность переносить кислород и более короткий срок жизни. Одна копия б не вызывает клинических проявлений, если не сочетается с другой мутацией гемоглобина, такой как HbC (b C) или бета-талассемия.
- Гемоглобин C: около 25% жителей Западной Африки и 2-3% афроамериканцев гетерозиготны по гемоглобину C (у них есть одна копия B C). Но заболевают только гомозиготные люди с обоими дефектными генами (b C). Обычные симптомы – легкая гемолитическая анемия с небольшим или средним увеличением селезенки.
- Гемоглобин E: вторая по распространенности гемоглобинопатия в мире с изменением бета-цепей. Патология очень часто встречается в Юго-Восточной Азии, особенно в Камбодже, Лаосе и Таиланде, а также частично в Северо-Восточной Азии. Есть случаи и в нашей стране. Люди с гомозиготным Hb E (две копии b E) обычно имеют легкую гемолитическую анемию, микроциты (маленькие красные кровяные тельца) и слегка увеличенную селезенку. Одна копия гемоглобина E не вызывает клинических признаков, если не сочетается с другой мутацией, такой как одна из бета-талассемии.
Талассемия
Необычные гемоглобинопатии
Существует ряд гемоглобинопатий, некоторые из которых не проявляются – они не вызывают никаких клинических признаков или симптомов. Другие, в свою очередь, влияют на функциональность и / или стабильность молекулы гемоглобина. Примерами являются гемоглобин D, гемоглобин G, гемоглобин J, гемоглобин M и гемоглобин Constant Spring. Мутации в гене альфа-цепи глобина приводят к образованию аномально длинных альфа (а) цепей, которые вызывают нестабильность в молекуле гемоглобина.
Другие примеры мутаций бета-цепи:
- Гемоглобин F: Hb F в основном вырабатывается в организме будущего ребенка (плода), и его функция заключается в переносе кислорода в среде с низким содержанием кислорода. Продукция гемоглобина F снижается сразу после рождения и стабилизируется на уровне взрослого человека до 1-2 лет. Гемоглобин F может быть повышен при некоторых врожденных заболеваниях. При бета-талассемии его уровень может быть нормальным или повышенным, но часто повышен при серповидно-клеточной анемии и сочетании серповидно-клеточной анемии с бета-талассемией. Пациенты с серповидно-клеточной анемией и повышенным Hb F часто имеют более легкое течение болезни, поскольку Hb F предотвращает серповидное движение красных кровяных телец. Уровни Hb F повышены в редком состоянии, называемом врожденным постоянством выработки гемоглобина плода (HPFH). Люди с повышенным уровнем гемоглобина F не имеют клинических признаков. HPFH вызывается разными генными мутациями у разных этнических групп. Hb F также может быть повышен при некоторых приобретенных состояниях, влияющих на выработку красных кровяных телец. Например, лейкемия и миелопролиферативные заболевания часто сопровождаются небольшим повышением уровня гемоглобина F.
- Гемоглобин H: HbH – это аномальный гемоглобин, который возникает в некоторых случаях альфа-талассемии. Его образование является ответом на фундаментальный недостаток альфа (а) цепей. Hb H состоит из четырех цепей бета (b) глобина. Хотя каждая из цепей бета-глобина нормальна, комплекс из четырех цепей бета нормально не функционирует. Обладает повышенным сродством к кислороду, плохо выделяет кислород клеткам тканей. Присутствие гемоглобина H также связано со значительным распадом эритроцитов (гемолизом), который возникает в результате осаждения нестабильного гемоглобина внутри красных кровяных телец.
- Hemoglobin Barts: Hb Barts вырабатывается в организме будущего ребенка с альфа-талассемией при условии, что все четыре гена, отвечающие за производство гемоглобина альфа, отсутствуют. Таким образом, не может образовываться гемоглобин HbA, HbA 2 и HbF. Гемоглобин Бартс состоит из четырех гамма (g) цепей и имеет высокое сродство к кислороду. Это состояние несовместимо с жизнью и обычно приводит к внутриутробной гибели плода.
Некоторые люди могут унаследовать два гена с разными мутациями, каждый от одного из родителей. Таких людей называют двойными или смешанными гетерозиготами.
Обследование: лабораторные тесты
Исследование на гемоглобинопатию проводится в следующих случаях:
- Выявление гемоглобинопатий у бессимптомных родителей больных детей.
- Выявление гемоглобинопатий у пациента с необъяснимой анемией, микроцитозом и / или гипохромией. Анализ может быть выполнен как часть теста на анемию.
- Скрининг на гемоглобинопатии у новорожденных – только в США и некоторых регионах с повышенной заболеваемостью.
- Пренатальный скрининг проводится в некоторых регионах с высокой частотой гемоглобинопатий (особенно в Африке).
На результаты тестов на гемоглобинопатию может повлиять переливание крови. Поэтому после переливания крови, прежде чем сдать анализ, пациенту следует подождать несколько месяцев. Тем не менее пациентам с серповидно-клеточной анемией после переливания крови рекомендуется сдать анализ крови, чтобы увидеть, достаточно ли гемоглобина в крови, и снизить риск повреждения организма серповидными эритроцитами.
Обследование гемоглобинопатий основано на обнаружении и оценке «нормальности» эритроцитов и гемоглобина в эритроцитах, а также на исследовании конкретной мутации гена. Каждый из тестов является частью головоломки, предоставляющей важную информацию о том, какая гемоглобинопатия присутствует. Для проверки гемоглобинопатии используются следующие тесты:
- Анализ крови. Анализ крови дает быструю информацию о клетках, циркулирующих в крови. Помимо прочего, результаты анализа крови показывают, сколько красных кровяных телец (эритроцитов) содержится в крови, какого они размера и формы, а также сколько там гемоглобина. Размер эритроцита определяет средний объем эритроцитов (MCV). Обнаружение пониженного MCV (микроцитоз, наличие небольших эритроцитов) часто сначала указывает на возникновение талассемии. Если MCV низкий и дефицит железа исключен, пациенты могут быть носителями талассемии или гемоглобинопатии, которые также вызывают микроцитоз (например, HbE).
- Анализ ДНК. Этот анализ используется для скрининга мутаций и делеций в альфа- и бета-областях глобиновых генов. Иногда обследуются все члены семьи. Задача в том, чтобы определить конкретный тип мутации, встречающейся в семье, и выявить всех носителей. ДНК-тесты не являются обычным тестом, но они могут помочь диагностировать гемоглобинопатию и выявить носителей.
- Мазок периферической крови (микроскопический дифференциальный подсчет лейкоцитов, считываемый по мазку периферической крови). Тест проводится путем формирования тонкого слоя крови на предметном стекле и окрашивания его специальными красителями. Образец крови, обработанный таким образом, затем оценивается лаборантом под микроскопом. Специалист определяет количество и тип белых и красных кровяных телец и тромбоцитов. Оценивает, являются ли они нормальными и зрелыми.
Анализ крови
При гемоглобинопатии эритроциты могут быть в следующих формах:
- Микроциты (меньше нормального).
- Гипохромные (более бледные, с пониженным гемоглобином).
- Разных размеров (анизоцитоз) и формы (пойкилоцитоз, например, серповидно-клеточные клетки).
- С ядром (в незрелых эритроцитах) или с включениями.
- С неравномерным распределением гемоглобина (клетки-мишени, которые под микроскопом выглядят как «бычий глаз»).
Наличие более высокого процента аномально выглядящих эритроцитов означает более высокую вероятность наличия заболевания.
С помощью тестов на гемоглобинопатию и их комбинаций можно диагностировать наиболее распространенные гемоглобинопатии. Эти тесты могут помочь выявить пациентов с сочетанием различных гемоглобинопатий (смешанные гетерозиготы).
Лечение гемоглобинопатии
В настоящее время гемоглобинопатии – неизлечимые заболевания. Но возможно устранять симптомы заболевания. Цель – облегчить боль и минимизировать возможные осложнения. Также существуют лекарства, повышающие уровень гемоглобина F, что облегчает некоторые симптомы.
Однако исследования и поиск более безопасных и эффективных методов лечения все еще продолжается. В будущем для восстановления мутированного гена можно будет использовать трансплантацию стволовых клеток или генную терапию. Для того чтобы эти методы могли широко использоваться в будущем, необходимы дальнейшие обширные исследования.
Источники: БЕРТИС, Калифорния, ЭШВУД, Эр., Брунс, Делавэр, (ред.), Учебник Тиц по клинической химии и молекулярной диагностике. 4-е издание Луи: Эльзевье-Сондерс, 2006; LOTHAR, T. Клиническая лабораторная диагностика. Франкфурт: TH-Books, 1998; MASOPUST, J. Клиническая биохимия – требования и оценка биохимических исследований, часть I. и часть 2, Прага: Каролинум, 1998; RACEK, J., et al. Клиническая биохимия. 2. переработанное издание, Прага: Гален, 2006; Каспер Д.Л., Браунвальд Э., Фаучи А.С., Хаузер С.Л., Лонго Д.Л., редакторы Джеймсон Д.Л., 2005.