Наследственные гемолитические анемии патофизиология

Мембранопатии
К мембранопатиям относят гемолитические анемии, обусловленные генетическим дефектом белковой (белковозависимые мембранопатии) или липидной (липидозависимые мембранопатии) компоненты цитоплазматической мембраны эритроцитов.
К белковозависимой мембранопатии относится наследственная микросфероцитарная анемия Минковского-Шоффара. В основе заболевания лежит дефект генов, кодирующих мембранные белки цитоскелета эритроцитов.
Эритроциты при болезни Минковского-Шоффара содержат аномальные формы спектрина, анкирина и белков, принимающих участие в поддержании нормальной двояковогнутой формы эритроцитов. В результате эритроциты лишаются возможности удерживать свою обычную форму, приобретают сферическую форму, теряют пластичность, способность к деформации, что нарушает их прохождение через узкие капилляры и особенно через синусы селезенки. Эритроциты в них задерживаются, что способствует их частичному разрушению макрофагами. Они теряют часть своей поверхности, уменьшаются в размере и превращаются в микросфероциты. Кроме того, дефектная мембрана эритроцитов становится высокопроницаемой для ионов натрия и воды, удаление избытка которых требует больших энергетических затрат, что приводит к сокращению срока их жизни со 120 до 12—14 дней.
Основными симптомами заболевания являются бледность, желтуха, спленомегалия, которые проявляются обычно с детства. Заболевание протекает с периодами ремиссии и обострения вплоть до гемолитического криза. Развитие обострения болезни связано с действием провоцирующих факторов, таких как психические переживания, физические нагрузки, инфекции. Выраженность анемии и клинических проявлений зависит от периода заболевания.
Для микросфероцитарной анемии характерны следующие изменения в периферической крови. Содержание гемоглобина и количество эритроцитов снижены, цветовой показатель в пределах нормы. В мазке отмечается микросфероцитоз — большое количество (до 40% всех эритроцитов) мелких круглых эритроцитов без центрального просветления, свойственного нормальным эритроцитам. В ответ на разрушение эритроцитов в костном мозге стимулируется эритропоэз с выходом в кровь большого количества ретикулоцитов. Содержание ретикулоцитов зависит от выраженности и периода заболевания и колеблется от 8-10 до 50—60% (при гемолитическом кризе). Количество лейкоцитов и тромбоцитов, как правило, нормальное. В период кризов может наблюдаться лейкоцитоз.
Дефекты белков мембраны эритроцитов могут приводить и к другим формам гемолитической анемии — наследственному эллипто- цитозу, стоматоцитозу и др. Это редко встречающиеся варианты гемолитической анемии.
Другой вид мембранопатии обусловлен появлением аномальных липидов в составе мембраны эритроцитов. Это приводит к своеобразным морфологическим изменениям в эритроцитах. В таких эритроцитах появляются выпячивания мембраны различного размера, расположенные на разных расстояниях друг от друга по поверхности клетки, в результате клетки становятся похожими на листья травянистого растения аканта, поэтому их называют акантоцитами. Они имеют и другое название — шпоровидные клетки. Низкая деформируемость, пониженная резистентность таких эритроцитов к различным воздействиям (изменению осмотического давления, температурным колебаниям, механическим факторам) являются причиной их повышенного распада и развития анемии.
Подобные изменения эритроцитов встречаются, в частности, у пациентов с наследственной абеталипопротеинемией.
Необходимо заметить, что эритроциты такой формы не являются строго специфичными для наследственного акантоцитоза. Они могут встречаться также при циррозе печени, в связи с нарушением липопротеинового обмена, при авитаминозе Е, у лиц с удаленной селезенкой.
Ферментопатии
В настоящее время известно более 20 наследственных энзимопа- тий эритроцитов, которые приводят к снижению продолжительности их жизни и повышенному гемолизу. К ним относятся нарушения активности ферментов гликолиза, пентозофосфатного цикла, системы глютатиона, метаболизма адениннуклеотидов и др. Из всех ферментопатий чаще всего встречаются гемолитические анемии, связанные с дефицитом ферментов пируваткиназы и особенно Г-6- ФДГ. Считается, что приблизительно 1/20 человечества имеет дефект фермента Г-6-ФДГ. Дефицит Г-6-ФДГ особенно отмечается в районах распространения малярии.
Гемолитическая анемия, обусловленная недостаточностью Г-6-ФДГ.
Известно большое количество мутантных форм Г-6-ФДГ (по данным разных авторов от 90 до 250), из которых две являются основными: более легкая африканская форма — тип А и более тяжелая средиземноморская форма — тип В. Последняя характеризуется не только снижением активности Г-6-ФДГ, как это имеет место при африканской форме, но и уменьшением его количества в эритроцитах.
Наследование дефицита Г-6-ФДГ сцеплено с Х-хромосомой, поэтому среди заболевших лиц преобладают мужчины.
При недостаточной активности Г-6-ФДГ нарушается пентозо- фосфатный цикл в эритроцитах, в связи с чем в недостаточном количестве синтезируется восстановленный кофермент НАДФН и не происходит образования восстановленного глютатиона. Восстановленный глютатион — необходимый компонент антиоксидантной системы эритроцитов. Он осуществляет защиту тиоловых групп гемоглобина и мембрану эритроцитов, прежде всего их липидов, от активных форм кислорода, образующихся в процессе жизнедеятельности самого эритроцита, а также от различного рода окислителей, возникающих при инфекциях, приеме определенных лекарственных препаратов, являющихся сильными окислителями (антималярий- ных, противотуберкулезных, сульфаниламидов, нитрофуранов, анальгетиков и пр.), при употреблении в пищу бобов vicia vafa. Эритроциты с пониженным содержанием глютатиона легко подвергаются действию окисляющих веществ. Образующиеся активные формы кислорода вызывают образование гидроперекисей ненасыщенных жирных кислот фосфолипидов, входящих в состав клеточных мембран, их разрушение и повышение проницаемости мембран эритроцитов для натрия и воды, что способствует их гемолизу. В результате действия гидроперекисей происходят также преципитация гемоглобина и отложение его в эритроцитах в виде телец Гейнца (округлых преципитатов числом более четырех). Это вызывает нарушение пластичности эритроцитов и снижение их способности к деформации при прохождении через капилляры.
зю
Заболевание, связанное с дефицитом ферментов Г-6-ФДГ, характеризуется желтушностью кожных покровов, умеренной нормохром- ной анемией, выраженность которых нарастает при переутомлении, инфекционных заболеваниях. Эти явления отмечаются с детских лет. Прием определенных лекарств-окислителей может привести к резкому обострению анемии. Гемолиз наступает не сразу после приема препарата, а спустя 2—3 дня. Возникает желтушное окрашивание кожи и слизистых оболочек, за счет гемоглобинурии моча приобретает темный цвет. Развивается тяжелая анемия, сопровождающаяся резким падением количества эритроцитов, снижением содержания гемоглобина (40—60 г/л), ретикулоцитозом, появлением в эритроцитах телец Гейнца. Форма эритроцитов самая разнообразная — от единичных микросфероцитов до мишеневидных макроцитов.
Гемолитическая анемия с дефектом пируваткиназы. Основным источником энергии в эритроцитах является гликолиз, поэтому снижение активности гликолитического фермента пируваткиназы тормозит производство энергии и снижает выработку АТФ. Дефицит энергии приводит к нарушению работы мембранных насосов, изменению ионного состава эритроцитов, укорочению продолжительности их жизни. Разрушение эритроцитов осуществляется главным образом внутриклеточно макрофагами селезенки и печени.
Дефицит пируваткиназы обнаруживается практически только у детей, у которых выраженный гемолиз имеется с самого рождения.
Гемоглобинопатии (гемоглобинозы)
Это состояние, вызванное мутацией генов, контролирующих синтез цепей глобина. Обычно эта патология встречается в странах с жарким климатом, эндемичных по малярии (в странах Африки, Средиземноморья, в Индии, Пакистане), реже в Средней Азии, на Кавказе.
В настоящее время описано более 200 аномальных типов гемоглобина, ведущих к гемоглобинопатиям. Правда, не все аномальные гемоглобины приводят к появлению патологических явлений. Некоторые же приводят к патологии, степень которой может быть различной — от небольших клинических проявлений, возникающих только при определенных условиях, до тяжелых, смертельных форм при гомозиготном наследовании мутантного гена.
Различают две группы наследственных гемоглобинопатий. Одна группа обусловлена наследственным дефектом первичной структуры цепей глобина, когда одна аминокислота в белковой цепи гемоглобина замещается другой. Вторая группа обусловлена наследственным дефектом синтеза некоторых полипептидных цепей глобина и заменой их другими.
Наследственные гемоглобинопатии относятся к числу наиболее распространенных в человеческой популяции генетических аномалий. Среди известных форм гемоглобинопатий наибольшее значение в клинической практике имеют серповидноклеточная анемия (ге- моглобиноз S) и талассемии.
Серповидноклеточная анемия (гемоглобиноз S). Нормальный гемоглобин взрослого человека состоит из НЬА (96%), НЬА2 (2,5%) и HbF (1,5%). Серповидноклеточная анемия — вид гемоглобинопатии, при котором появляется аномальная форма гемоглобина — HbS (от англ, sikle — серп). Эта форма гемоглобинопатии получила такое название, поскольку гемоглобин при определенных условиях вызывает деформацию эритроцита и придает ему серповидную форму. Гемоглобин S образуется в результате точечной мутации в структурном гене, приводящей к замещению глутаминовой кислоты, стоящей в шестом положении p-цепи, валином. Такая замена уменьшает растворимость гемоглобина. Отличительными особенностями HbS являются следующие: растворимость его восстановленной формы примерно в 100 раз ниже нормального НЬА, он приобретает способность к полимеризации и выпадению в осадок в виде кристаллов. Эритроциты с высокой концентрацией HbS при падении парциального давления кислорода в окружающей среде, при циркуляции в зоне гипоксии, ацидоза деформируются, приобретают форму серпа, повреждаются выпавшими острыми кристаллами.
Наследование аномального гена может быть гетерозиготным и гомозиготным. При гетерозиготном наследовании патологический гемоглобин S составляет обычно 20—45% всего гемоглобина клетки, а остальное количество приходится на нормальный гемоглобин А, поэтому у людей с такими нарушениями обычно развивается легкая форма заболевания, у 8—10% даже клинически не проявляющаяся. В случаях гомозиготного наследования в эритроцитах содержится 80—100% гемоглобина S, поэтому тяжелая анемия возникает у гомозиготных по HbS детей.
Изменение формы эритроцитов снижает их деформируемость, что затрудняет прохождение эритроцитов по микрососудам и способствует замедлению кровотока, развитию стаза в сосудах микро- циркуляторного русла. В результате возникает ишемическое повреждение различных тканей. Ишемическому повреждению тканей способствует закупорка сосудов продуктами гемолиза измененных эритроцитов. Средняя продолжительность жизни эритроцитов у больных, гомозиготных по HbS, составляет в среднем 17 сут.
В большинстве неосложненных случаев анемия при гемоглоби- нозе S умеренная (содержание гемоглобина — 100—80 г/л, эритроцитов — 3,5—3,0 • 1012/л), носит нормохромный характер. В мазке наряду с серповидными эритроцитами (или дрепаноцитами) выявляются мишеневидные эритроциты с базофильной пунктацией цитоплазмы (рис. 15.6) Содержание ретикулоцитов повышено. Отмечаются нейтрофил ьный лейкоцитоз, тромбоцитоз.
Помимо серповидноклеточной анемии известны формы гемоглобинопатий, характеризующиеся наличием других аномальных типов гемоглобина. Из этих аномалий чаще всего встречается гемоглобинопатия С. В гемоглобине С глутаминовая кислота в шестом положении (3-цепи замещена лизином, вследствие чего гемоглобин кристаллизуется, но при этом эритроциты приобретают не серповидную, а мишеневидную форму. Клинические проявления выявляются только при гомозиготном носительстве.
Рис. 15.6. Картина периферической крови при серповидноклеточной анемии (по Lippincott Williams-Wilkins, 2010)
Талассемии (средиземноморская анемия) — группа гемолитических анемий, в основе которых лежит угнетение синтеза одной из нормальных цепей глобина. В образовании молекулы глобина принимают участие четыре полипептидные цепи, обозначаемые буквами греческого алфавита, — альфа (а), бета ((3), гамма (у) и дельта (8). Основной гемоглобин взрослого человека НЬА состоит из двух a-цепей и двух (3-цепей; НЬА2 — из двух а-цепей и двух 8-цепей. В состав HbF входит по две а- и у-цепи. Таким образом, все типы гемоглобина состоят из двух a-цепей и двух цепей другого типа. Такое сочетание цепей в составе молекулы глобина позволяет гемоглобину сохранять высокую растворимость. Нарушение синтеза той или иной цепи глобина приводит к образованию нестабильных форм гемоглобина, их преципитации в эритробластах и в конечном итоге к ускоренному повреждению эритроцитов в циркулирующей крови.
Возможно полное или частичное угнетение синтеза любой цепи глобина, но чаще всего встречается патология а- и особенно (3-цепи, в связи с чем различают два основных типа талассемий: альфа и бета.
Альфа-талассемия. Спектр клинических проявлений заболевания весьма широк: от бессимптомного носительства мутантного гена до патологии, не совместимой с жизнью. В синтезе a-цепи глобина принимают участие четыре гена. Клиническая картина а-талассемии зависит от количества дефектных генов, ответственных за синтез a-цепи, и степени нарушения синтеза последней. Адекватное количество a-цепей способно образовываться до тех пор, пока не поражаются три или четыр а-глобиновых гена. В случаях полного угнетения синтеза a-цепи не образуются гемоглобины А, А2 и F. Избыточные у-цепи глобина принимают участие в формировании патологического гемоглобина Барта с очень высоким сродством к кислороду. Этот гемоглобин не отдает кислород в ткани плода, из-за чего возникают тканевая гипоксия и, как правило, внутриутробная смерть.
При мутации трех а-глобиновых генов образуется гемоглобин Н, состоящий из четырех (3-цепей, подобно гемоглобину Барта имеющий слишком высокое сродство к кислороду и легко выпадающий в осадок. Эритроциты, содержащие гемоглобин Н, характеризуются меньшей продолжительностью жизни и быстро разрушаются в селезенке. Это ведет к развитию умеренной микроциртарной анемии вследствие гемолиза эритроцитов.
Дисфункция одного или двух из четырех а-глобиновых генов либо клинически бессимптомна, либо вызывает незначительную микроцитарную гемолитическую анемию.
Бета-талассемия. Патологические изменения при (3-талассемии обусловлены нарушением синтеза (3-цепи глобина и образованием нестабильных гемоглобинов, содержащих только а-цепи.
По тяжести клинической картины (3-талассемии делят на большую, малую и минимальную. Большая талассемия (болезнь Кули) встречается у гомозиготов и характеризуется резким снижением синтеза НЪА при значительном компенсаторном увеличении синтеза HbF и НЬА2 — форм гемоглобинов с более высокой кислородосвязывающей способностью по сравнению с НЬА. Неэффективный эритропоэз из-за избыточного синтеза a-цепи, снижение кислородной емкости крови ведут к развитию тканевой гипоксии, интенсивной эритроидной гиперплазии и значительному расширению объема зон кроветворения, в том числе на все черепные кости. Гиперплазия костно-мозгового кроветворения в костях верхней челюсти приводит к типичному изменению лица: скулы выдаются, прикус зубов нарушается из-за того, что верхняя челюсть становится диспропорционально больше, чем нижняя, поэтому болезнь характеризуется не только прогрессирующей анемией с гепатоспленомегалией, повышенным гемолизом с урибилинурией, но и остеопорозом со своеобразными изменениями лицевого скелета по «монголоидному» типу.
В крови наблюдается тяжелая гипохромная, микроцитарная анемия, сопровождающаяся нормальным или повышенным содержанием железа сыворотки. В мазке резко выражен анизопойкилоцитоз, преобладают мишеневидные эритроциты, часто выявляется базо- фильная пунктация эритроцитов (рис. 15.7). Отмечается высокий периферический ретикулоцитоз. Характерна лейкопения с относительным лимфоцитозом и только в период криза возможен нейтро- фильный лейкоцитоз. Количество тромбоцитов нормальное или сниженное.

Рис. 15.7. Картина периферической крови при талассемии (по Lippincott Williams-Wilkins, 2010)
Малая и минимальная р-талассемии возникают в результате наследования дефектного гена лишь от одного из родителей. Болезнь протекает с нерезко выраженными клиническими проявлениями, иногда бессимптомно. Гематологические показатели при гетерозиготной талассемии могут варьировать в значительных пределах. Как правило, наблюдается микроцитарная гипохромная анемия. При исследовании мазка периферической крови можно обнаружить выраженный пойкилоцитоз — мишеневидные эритроциты, дакриоци- ты (каплеподобные клетки), эритроциты с базофильной зернистостью, ядерные эритроциты.
(для
внутрикафедрального пользования)
МИНИСТЕРСТВО
ЗДРАВООХРАНЕНИЯ РЕСПУБЛИКИ БЕЛАРУСЬ
УО
«ГОМЕЛЬСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ
УНИВЕРСИТЕТ»
Кафедра
патологической
физиологии
Утверждено
на заседании кафедры
протокол
№
7 от 31.08.2015
Зав.
кафедрой патофизиологии, к.м.н.
доцент_____________Т.С.
Угольник
Учебно-методическая
разработка для студентов
лечебного
факультета
Гомель
2015
Патофизиология системы крови. Гемолитические анемии. Эритроцитозы
Актуальность
темы: Гемолитические
анемии различны по этиологии, механизмам
развития, клинико-гематологической
картине, поэтому изучение этих видов
анемий особенно важно в практической
деятельности врача.
2.
Цель занятия:
изучить этиологию, патогенез и основные
проявления гемолитических анемий.
3.
Задачи занятия: Знать
классификацию, этиологию и патогенез
гемолитических анемий. Изучить основные
нарушения и компенсаторно-приспособительные
процессы в организме при анемиях и
полицитемиях.
Основные учебные вопросы (план):
Патология
системы эритрона. Анемии и эритроцитозы,
определение понятий, принципы
классификации, общая характеристика.Анемии
при лейкозах и других опухолевых
процессах.Наследственные
гемолитические анемии: эритроцитопатии,
эритроэнзимопатии, гемогобинопатии
причины и механизмы развития.Приобретенные
гемолитические анемии: виды, причины,
механизмы развития, проявления в
органах кроветворения и в периферической
крови.Роль
аутоиммунных процессов в патогенезе
анемий. Аутоиммунные гемолитические
анемии.Эритроцитозы
первичные и вторичные: причины, механизмы
развития, проявления в органах
кроветворения и в периферической
крови.Нарушения
и компенсаторно-приспособительные
процессы в организме при анемиях и
эритроцитозах.Принципы
терапии анемий.
5. Вспомогательные материалы по теме:
Группа
анемий, наследственно обусловленных
или приобретенных, общим признаком
которых является укорочение жизни
эритроцитов. При этом имеет место стойкое
(хроническая ГА) или массированное
(острая ГА) преобладание разрушения
эритроцитов над их образованием.
Проявляется заболевание синдромами
усиленного гемолиза и компенсаторного
усиления эритропоэза. Усиление гемолиза
(гемолитические кризы) наблюдается при
всех ГА и нередко развивается после
интеркуррентных заболеваний, большой
физической нагрузки, в результате
стрессов, интоксикаций и т. д. В ряде
случаев провоцирующий агент установить
не удается.
Классификация га:
I. Наследственные:
– эритроцитопатии
(мембранопатии);
– ферментопатии
(энзимопатии);
– гемоглобинопатии
(гемоглобинозы).
II. Приобретенные:
Иммунные
формы:
изоиммунные
гетероиммунные
формыаутоиммунные
формы
Неиммунные
формы:
токсико-гемолитические
инфекционные
механические
Развитие
наследственных ГА обусловлено внутренними
аномалиями эритроцитов (эндоэритроцитарные);
приобретенных — влиянием факторов,
действующих вне эритроцита
(экзоэритроцитарные).
Гемолиз
эритроцитов
при гемолитических анемиях может
происходить внутриклеточно (так же как
и физиологический гемолиз), или
непосредственно в сосудах. В связи с
этим выделяют 2 типа патологического
гемолиза:
1.
Внутриклеточный гемолиз—
разрушение «маркированных» иммуноглобулином
(Ig) G эритроцитов в РЭС при наследственной
патологии мембраны эритроцитов,
нарушениях активности ферментов, синтеза
гемоглобина, при несовместимости по
эритроцитарным антигенам между матерью
и плодом и при гемотрансфузиях.
2.
Внутрисосудистый гемолиз
— комплементзависимый лизис «маркированных»
IgM (реже IgG) эритроцитов непосредственно
в кровотоке (в сосудах) при действии
каких-либо внешних факторов, которые
вызывают прямое или опосредованное
повреждение клеток. Причиной этого
может быть разрушение мембраны эритроцитов
вследствие механической травмы (при
окклюзии сосудов, гемодиализе, протезах
клапанов сердца и др.), под влиянием
физических (ионизирующая радиация,
высокая температура), токсических (при
действии экзо- и эндотоксинов), инфекционных
и иммунных (при образовании антиэритроцитарных
аутоантител) патологических факторов.
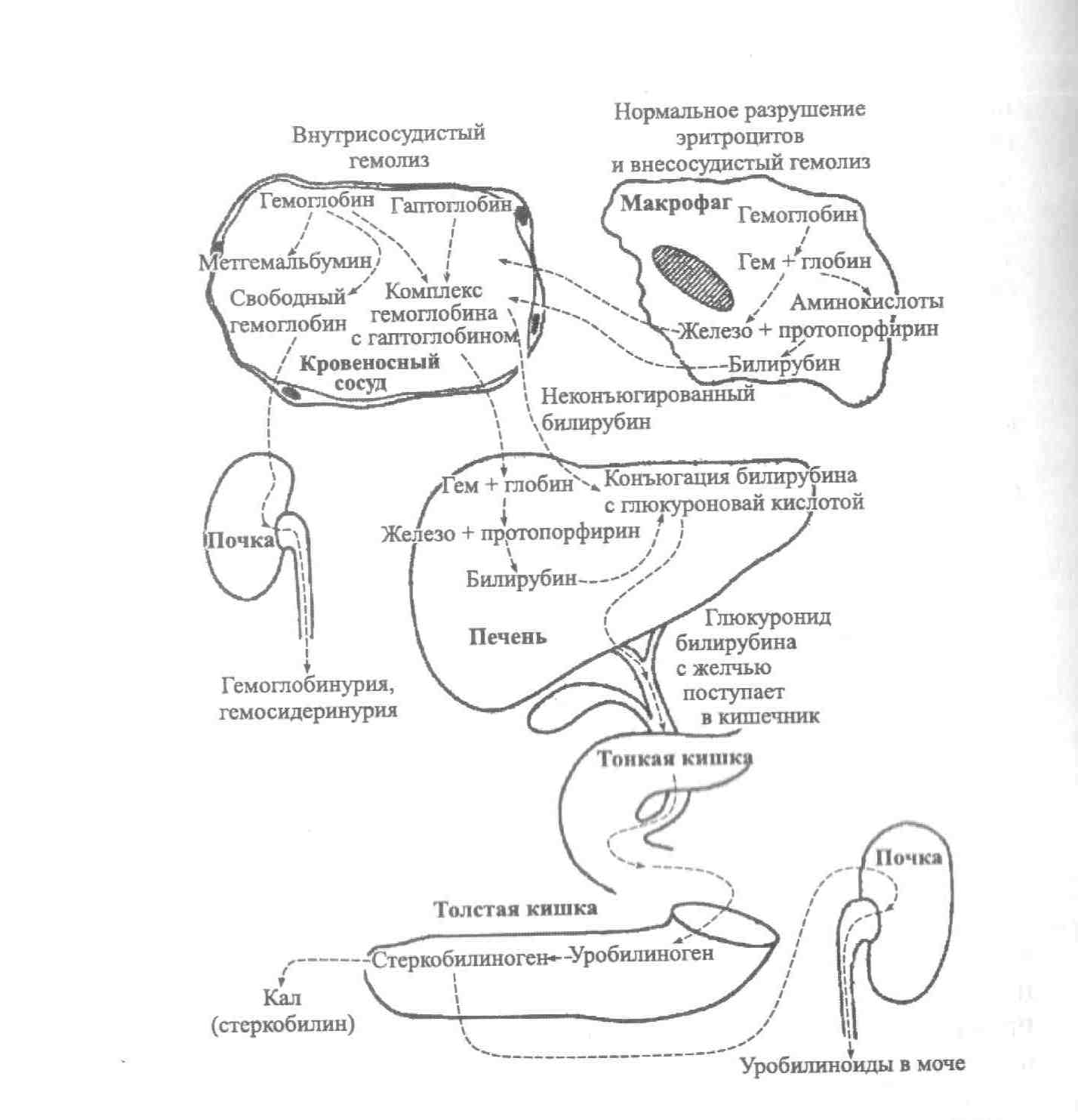
Рисунок
– 1. Механизмы внутрисосудистого и
внутриклеточного гемолиза.
Внутриклеточный
гемолиз
происходит внутри фагоцитов. Из
разрушенных эритроцитов освобождается
гемоглобин, который затем распадается
на глобин, железо и протопорфирин. Глобин
под действием протеолитических ферментов
расщепляется на аминокислоты, с плазмой
они переносятся во внутренние органы
для дальнейшего синтеза белков. Железо
при помощи трансферрина транспортируется
в кровь, затем в костный мозг и в органы,
депонирующие железо (преимущественно
в печень). Протопорфирин превращается
в биливердин, а затем в билирубин
(неконъюгированный билирубин), который
транспортируется альбумином в клетки
печени, где связывается с глюкуроновой
кислотой и метаболизируется в диглюкуронид.
Диглюкуронид выделяется в желчь, а затем
в кишечник, где превращается в уробилиноген,
затем стеркобилиноген и выводится с
мочой (уробилиноиды) и калом.
При
внутрисосудистом
гемолизе
эритроциты распадаются в кровеносном
русле, высвобожденный из эритроцитов
гемоглобин связывается с гаптоглобином
плазмы и транспортируется к клеткам
ретикулоэндотелиальной системы печени,
где происходит дальнейший распад
гемоглобина с образованием свободного
билирубина, который затем подвергается
глюкуронидированию. Не связавшийся с
гаптоглобином гемоглобин выводится с
мочой (гемоглобинурия, гемосидеринурия).
В
результате повышенного гемолиза
эритроцитов в крови накапливается
большое количество непрямого билирубина,
что приводит к развитию желтухи. Помимо
этого главным признаком повышенного
внутриклеточного гемолиза является
увеличение селезенки (спленомегалия),
в случаях внутрисосудистого разрушения
эритроцитов ведущим симптомом становится
появление гемоглобина в моче
(гемоглобинурия), что сопровождается
изменением ее окраски вплоть до черного
цвета (табл. 1).
Таблица
1.
Дифференциальные
признаки внутрисосудистого и
внутриклеточного гемолиза
Признаки | Виды | |
внутрисосудистый | внутриклеточный | |
Локализация | Сосуды | РЭС |
Локализация | Канальцы | Селезёнка, |
Желтушность | Умеренная | Выраженная |
Увеличение | Незначительное | Значительное |
Ведущие | Нормохромная | |
Гемоглобинемия Гемоглобинурия Гемосидеринурия | Гипербилирубинемия Повышенное | |
Все
формы малокровия, связанные с повышенной
гибелью эритроцитов периферической
крови, относятся к группе регенераторных
анемий с нормобластическим типом
эритропоэза.