Норма муковисцидоза в крови

Общий анализ крови
Общий анализ крови (ОАК) – это лабораторный метод диагностики, которой подразумевает:
- Подсчет кровяных телец всех типов (красных – эритроцитов, белых – лейкоцитов и тромбоцитов) и исследование их характеристик (размеры, форма и др.),
- Составление лейкоцитарной формулы (процентное соотношения количества лейкоцитов всех форм).
- Определение количества гемоглобина (Hb) в одной единице объема.
- Составление соотношения массы всех форменные элементов крови к ее плазме (гематокрит).
Таблица услуг
| Название услуги | Цена |
|---|---|
| Акция! Первичная консультация репродуктолога и УЗИ | 0 руб. |
| Первичная консультация репродуктолога, к.м.н. Осиной Е.А. | 10 000 руб. |
| Консультация генетика по скайпу или телефону | 2 500 руб. |
| Повторная консультация генетика | 2 500 руб. |
| Первичная консультация генетика (Консультация генетика – выявление риска рождения ребенка с наследственными заболеваниями) | 3 500 руб. |
| Консультация генетика перед ПГД | 3 300 руб. |
| Тест Vistara (Natera) | 78 000 руб. |
| Тест Vistara совместно с Panorama базовой (Natera) | 95 550 руб. |
| Тест Vistara совместно с Panorama расширенной (Natera) | 107 250 руб. |
| Неинвазивное пренатальное исследование плода на наличие анеуплодий (PANORAMA) | 33 900 руб. |
| Неинвазивное пренатальное исследование плода на наличие анеуплоидий + панель микроделеций (PANORAMA) | 47 500 руб. |
| Цитогенетическое исследование материала абортуса (24 хромосомы, aGGH) | 15 000 руб. |
| Тест ANORA цитогенетический анализ абортуса ( Natera) | 53 200 руб. |
| Пренатальный комплекс 10-13 нед. (свободная b-субъединица хорионического гонадотропина человека (свободный b-ХГЧ, free b-hCG) | 2 100 руб. |
| Пренатальный комплекс 14-20 нед. (Хорионический гонадотропин человека (ХГЧ, бета-ХГЧ, б-ХГЧ, Human Chorionic gonadotropin, HCG) | 2 400 руб. |
| Панель Horizon 4 (Natera) | 27 300 руб. |
| Панель Horizon 27 (Natera) | 31 200 руб. |
| Панель Horizon 106 (Natera) | 42 900 руб. |
| Панель Horizon 274 (Natera) | 50 700 руб. |
| Предварительный этап ПГТ для носителей моногенных мутаций (кровь с ЭДТА) | 112 500 руб. |
| ПГТ моногенные заболевания (до 8 эмбрионов) | 187 000 руб. |
| ПГТ на 24 хромосомы при проведении ПГТ моногенного заболевания (1 эмбрион) | 20 000 руб. |
| Преимплантационное генетическое тестирование на 24 хромосомы (1 эмбрион) бесплатная консультация генетика по результатам ПГТ | 23 000 руб. |
| Преимплантационная генетическая экспресс диагностика на 24 хромосомы (1 эмбрион) | 48 000 руб. |
| Преимплантационное генетическое тестирование на 24 хромосомы – за каждый дополнительный эмбрион, начиная с 3-го | 20 000 руб. |
| Преимплантационное генетическое тестирование 24 хромосомы + транслокация (до 2 эмбрионов) | 72 000 руб. |
| Преимплантационное генетическое тестирование 24 хромосомы + транслокация (за каждый дополнительный эмбрион, начиная с 3) | 35 000 руб. |
| Преимплантационное генетическое тестирование эмбрионов, остановившихся в развитии | 60 000 руб. |
| Подготовка эмбрионов для повторного генетического исследования | 10 780 руб. |
Получить бесплатную консультацию врача
Результаты общего анализа крови при муковисцидозе
В общем анализе крови больного с муковисцидозом можно определить неспецифические отклонения он нормы. К ним относятся: общее снижение количества эритроцитов, уровня Hb и количества лейкоцитов.
При развитии осложнений, в виде гнойно-обструктивного бронхита или пневмонии показатели общего анализа крови меняются: возрастает уровень лейкоцитов, скорость оседания эритроцитов (СОЭ).
Скрининг диагностика – иммунореактивный трипсиноген
Для выявления скрытых форм данной патологии у детей, в том числе и младенцев, используются специальные скрининговые тесты. В какой-то мере этот метод диагностики равноценен определению трипсина в кале, однако в этом случае для постановки диагноза «Муковисцидоз» используется анализа крови.
Суть этого метода состоит во взятии небольшого количества крови ребенка и измерения в нем уровня фермента пищеварительной системы – иммунореактивного трипсиногена. Данный энзим присутствует и у здоровых детей, однако у детей больных на муковисцидоз его уровень значительно выше.
Нормальные показатели иммунореактивного трипсиногена у детей:
| Возраст, годы | Иммунореактивный трипсиноген, мкг/л |
|---|---|
| При рождении | 23.3 ± 1.9 |
| 0 – ½ | 31.3 ± 5.4 |
| ½ – 1 | 37.1 ± 6.9 |
| 1 – 3 | 29.8 ± 1.8 |
| 3 – 5 | 28.3 ± 3.2 |
| 5 – 7 | 35.7 ± 3.6 |
| 7 – 10 | 34.9 ± 2.2 |
Генетический анализ при муковисцидозе
Верификация диагноза происходит по итогам такого исследования, как генетический анализ на муковисцидоз. Он выявляет различные варианты мутации (изменений) гена CFTR.
[42-032]
Генетическая диагностика муковисцидоза. Анализ гена CFTR (25 мутаций)
15165 руб.
Комплексное генетическое исследование, которое позволяет выявить 25 наиболее часто встречающихся на территории России мутаций гена CFTR, приводящих к развитию тяжелого наследственного заболевания муковисцидоза.
Метод исследования
БиоЧип.
Какой биоматериал можно использовать для исследования?
Венозную кровь, буккальный (щечный) эпителий.
Как правильно подготовиться к исследованию?
Специальной подготовки не требуется.
Общая информация об исследовании
Муковисцидоз (синоним – кистозный фиброз) – одно из наиболее распространенных аутосомно-рецессивных наследственных заболеваний человека. Он характеризуется нарушением функции эпителия дыхательных путей, кишечника, поджелудочной железы, потовых и половых желез.
Причиной развития муковисцидоза являются мутации в гене CFTR (cystic fibrosis transmembrane regulator), кодирующем АТФ-связывающий белок, который формирует канал для ионов хлора в клеточных стенках. Мутации приводят к нарушению транспорта электролитов и ионов хлора через мембраны эпителиальных клеток, что сопровождается усилением секреции густой слизи и закупоркой выводящих протоков различных желез.
Существует несколько форм муковисцидоза:
- смешанная (поражаются одновременно органы дыхания и пищеварительный тракт);
- бронхолегочная (поражаются преимущественно органы дыхания);
- кишечная (поражается преимущественно желудочно-кишечный тракт);
- мекониевая непроходимость кишечника;
- атипичные формы, связанные с изолированными поражениями отдельных желез внешней секреции.
В настоящее время в РФ диагноз “муковисцидоз” ставится одному из 9 000 новорождённых (для сравнения: в Европе муковисцидоз диагностируется с частотой 1 : 2 000 – 3 000 новорождённых). Однако принятая в России форма массового скрининга новорождённых несовершенна и иногда не позволяет выявить заболевание на доклинической стадии.
В каждой клетке нашего организма имеется две копии гена CFTR. Одна копия достается от отца, а вторая от матери. Заболевание муковисцидоз аутосомно-рецессивное, т. е. развивается оно только при условии, что ребенок получает и от отца, и от матери мутантный ген CFTR. При этом родители, у которых вторые копии гена CFTR нормальные, не страдают муковисцидозом и порой даже не догадываются о его носительстве. По статистике, в европейской популяции носителем мутаций гена CFTR в среднем является каждый 25-й человек.
Насчитывается примерно одна тысяча различных мутаций в гене CFTR. Они встречаются с различной частотой в разных популяциях. Некоторые нарушения в гене могут не иметь никаких проявлений. Но большая часть мутаций вызывает патологический эффект, т. к. приводит к нарушению функционирования белка.
В данное комплексное исследование включен анализ гена CFTR на 25 мутаций, наиболее распространенных на территории РФ, Восточной Европы и Скандинавии и связанных с развитием тяжелых клинических форм муковисцидоза. Исследование позволяет выявлять до 95 % всех возможных больных, что существенно превышает разрешающие способности утвержденного в России скрининга.
Исследование поможет не только подтвердить или опровергнуть диагноз “муковисцидоз”, но и выявить носительство мутации у здоровых людей. Особенно важно проводить генетическое тестирование в семьях, в которых есть больные муковисцидозом, поскольку у пары, где оба родителя являются носителями мутаций, вероятность рождения больного ребенка составляет 25 %.
До сих пор муковисцидоз считается неизлечимым заболеванием, но ранняя диагностика и адекватная терапия значительно улучшают прогноз заболевания и продляют пациенту жизнь.
Перечень исследуемых мутаций в гене CFTR:
Dele2-3 | R347H | G542X | 2184insA | 3732delA |
G85E | 1078delT | G551D | 2183AA-G | 3821delT |
621+1G>T | I507del | R553X | 2789+5G>A | 3849+10kbC>T |
R334W | F508del | 1717-1G>A | R1162X | W1282X |
R347P | 1677delTA | 2143delT | S1196X | N1303K |
Когда назначается исследование?
- Генетическая диагностика в рамках неонатального скрининга
- Клиническая молекулярно-генетическая диагностика для подтверждения диагноза, поставленного во взрослом возрасте
- Пренатальная диагностика в случае семейного анамнеза заболевания
- Определение риска рождения ребенка с муковисцидозом при планировании семьи
- Диагностика мужского бесплодия
Что означают результаты?
В ходе анализа проводится исследование 25 значимых генетических маркеров гена CFTR, что позволяет обнаружить наиболее распространенные мутации, приводящие к развитию заболевания.
- N (норма) / N (норма) – мутации не обнаружены.
- N / M (мутации) – выявлена гетерозиготная мутация, скрытое носительство.
- M (мутация) / M (мутация) – выявлена гомозиготная мутация, подтверждение диагноза “муковисцидоз”.
По итогам комплексного исследования выдается заключение врача-генетика с интерпретацией результатов.
Литература
- А. Е. Павлов, С. В. Апалько, Е. В. Воробьев. Молекулярно-генетическая диагностика муковисцидоза в формате микрочипа. Лаборатория №4, 2012.
- American College of Medical Genetics Laboratory standards and guidelines for CFTR Mutation Testing (2011).
Одним из важных методов ранней диагностики муковисцидоза, определяющий своевременное начало терапии, увеличение продолжительности и улучшению качества жизни больных, является массовый скрининг новорожденных. В мире неонатальный скрининг на муковисцидоз успешно проводится более тридцати лет. Как показал многолетний опыт, активное диспансерное наблюдение и своевременное комплексное лечение вновь выявленных больных, позволяют предотвратить или, по крайней мере, замедлить развитие осложнений.
По сравнению с больными, диагностированными по симптомам заболевания в более позднем возрасте, для выявленных при неонатальном скрининге характерно хорошее самочувствие. К трем годам отмечены достоверно меньшие изменения в легких, меньшая частота обострений бронхолегочного процесса, более редкая частота высева патогенной микрофлоры. Наблюдается также значимо меньшая частота декомпенсации кишечного синдрома, лучшие показатели физического статуса, включая массо-ростовой индекс.
С июня 2006 года в ряде регионов РФ, а с 2007 года по всей России проводится скрининг новорожденных на муковисцидоз. Обследование состоит из 4х этапов, три из которых обязательны.
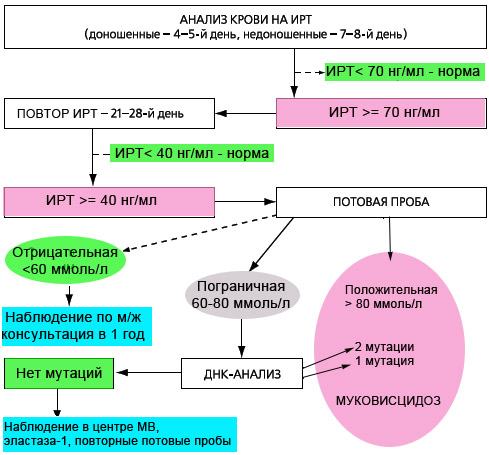
- Первый этап — проводится всем детям. У новорожденных берут каплю крови на 4-5й день жизни (у недоношенных на 7-8й день), и в высушенном пятне крови определяется содержание иммунореактивного трипсина (ИРТ). Нормальное значение: не более 65-70 нг/мл (в зависимости от используемых в данной лаборатории реактивов).
- Второй этап — у детей с повышенным уровнем ИРТ (>70 нг/мл) повторное определение ИРТ в крови проводят на 4 неделе жизни (с 21 по 28 день жизни строго). У здоровых детей показатель не превышает 40 нг/мл.
Повышение уровня ИРТ у новорожденных бывает не только при муковисцидозе, но и при ряде врожденных и наследственных патологий, таких как: внутриутробная гипоксия плода, внутриутробные инфекции, перинатальный стресс, незрелость плода, коньюгационная желтуха новорожденных, хромосомные перестройки и др., а также гетерозиготное носительство мутаций в гене CFTR, как следствие функциональной недостаточности поджелудочной железы. Поэтому для подтверждения диагноза МВ при положительных результатах 2го теста ИРТ (>40 нг/мл), а также при несоблюдении сроков проведения 2го теста (21-28 день) необходимо обязательно проводить потовую пробу. И наоборот, при некоторых состояниях (у недоношенных детей, при мекониальном илеусе, при вирусных инфекциях, после переливаний крови) возможно получение ложноотрицательных результатов. То есть диагноз МВ впоследствии может быть поставлен и при отрицательных результатах скрининга.
- Третий этап. Потовая проба, проводится детям с положительными результатами определения ИРТ. Нормальные значения: до 50 ммоль/мл.
Если результаты потовой пробы не превышают 50 ммоль/л, за развитием ребенка наблюдают по месту жительства, повторная консультация проводится в 1 год.
- Четвертый этап. При пограничном результате потовой пробы (40-60 ммоль/л по методу Гибсона-Кука или 60-80 ммоль/л (Нанодакт)) проводится ДНК-диагностика на 20 наиболее распространенных в РФ мутаций МВ.
Если мутаций не выявлено, то ребенок в течение года наблюдается в ближайшем центре муковисцидоза, необходимо провести исследование содержания эластазы 1 в кале, провести повторные потовые пробы.
При положительном результате потовой пробы (>60 ммоль/л по методу Гибсона-Кука или >80 ммоль/л при определении проводимости пота на аппарате Нанодакт) диагноз МВ считается подтвержденным. Ребенок наблюдается в центре муковисцидоза.
Литература:
1. В.Д. Толстова, Н.Ю. Каширская, Н.И. Капранов. Массовый скрининг новорожденных на муковисцидоз в России. Фарматека № 1 — 2008

Обследование новорожденного проводится амбулаторно с соблюдением мер по предупреждению перекрестного инфицирования. Госпитализация – в случае развития тяжелого обострения, требующего мониторинга состояния и проведения в/в терапии. При госпитализации необходимо соблюдать принцип разделения больных по характеру высеваемой микрофлоры, оптимально – госпитализировать в боксы (Приказ МЗ РФ от 15 мая 2012 г. № 535н «Об утверждении перечня медицинских и эпидемиологических показаний к размещению пациентов в маломестных палатах (боксах)» https://legalacts.ru/doc/prikaz-minzdravsotsrazvitija-rossii-ot-15052012-n-535n/).
Повторная консультация после постановки диагноза должна быть проведена не позднее 2-х недель, по желанию родителей – раньше. Родители ребенка с МВ должны иметь возможность консультироваться со специалистом по мере возникновения такой необходимости (в рабочие часы). Они должны иметь инструкции, куда обращаться в нерабочие часы в случае непредвиденных обстоятельств [9].
Частота осмотров: до 3-х мес – каждые 2 недели, 3-6 мес – ежемесячно, 6-12 мес – 1 раз в 2 мес, далее – ежеквартально.
С момента постановки диагноза «МВ» ребенок должен наблюдаться командой специалистов: врач-педиатр (специалист по муковисцидозу), кинезитерапевт, нутрициолог, а семья должна иметь возможность получать консультации психолога, врача-генетика, сибсам должна быть проведена потовая проба. При необходимости привлекаются врачи других специальностей.
В случае невозможности проведения в короткие сроки подтверждающей диагностики МВ (потовый тест, ДНК-диагностика), при наличии характерных клинических проявлений заболевания (кишечный синдром со стеатореей, задержка физического развития, респираторные проявления, мекониевый илеус и др.) диагноз «МВ» может быть установлен клинически. Незамедлительно должна быть начата посиндромная терапия (заместительная ферментная, муколитическая, терапия жирорастворимыми витаминами, добавление соли в пищу). Подтверждающая диагностика в этих случаях может быть проведена позднее.
II. Диагностика по клиническим признакам
Диагностика классической формы МВ обычно не представляет сложностей. Классический фенотип больного является результатом наличия двух мутантных копий гена муковисцидозного трансмембранного регулятора (CFTR), имеющих клинические последствия (https://seqdb.med-gen.ru/), и характеризуется хронической бактериальной инфекцией дыхательных путей и придаточных пазух носа, стеатореей вследствие внешнесекреторной недостаточности поджелудочной железы, мужским бесплодием из-за обструктивной азооспермии, а также повышенной концентрацией хлоридов потовой жидкости [1, 3, 4, 6, 29]. Проблемы диагностики МВ, как правило, связаны с фенотипическим разнообразием его форм, обусловленным генетическим полиморфизмом заболевания, наряду с влиянием генов-модификаторов, факторов внешней среды (медикаментов, поллютантов, курения и др.). Пациенты с так называемым атипичным муковисцидозом имеют как минимум одну копию мутантного гена CFTR, функция которого частично сохранена, – «мягкие» мутации. В ряде случаев «мягкие» мутации МВ обусловливают его диагностику во взрослом возрасте. Как правило, в этой группе больных отмечается более мягкое течение болезни в связи с сохранностью функции поджелудочной железы и нетяжелым поражением органов дыхания [37].
В абсолютном большинстве случаев МВ может быть диагностирован в раннем детском возрасте (в 90% случаев – на первом году жизни). К сожалению, нередки случаи диагностики МВ у взрослых с классическим фенотипом.
Учитывая возможность получения ложноотрицательных результатов неонатального скрининга, а также то обстоятельство, что в РФ неонатальный скрининг на МВ проводится с 2006–2007 гг., не теряет своей актуальности анализ групп риска, включающих пациентов с патологией желудочно-кишечного тракта, бронхолегочными нарушениями, патологией других органов, а также родственников больных МВ (Табл. 3).
Таблица 3. Группы риска для дифференциальной диагностики муковисцидоза [1]
I. Бронхолегочные нарушения
1. Повторные и рецидивирующие пневмонии с затяжным течением, особенно двусторонние
2. Бронхиальная астма, рефрактерная к традиционной терапии
3. Рецидивирующие бронхиты, бронхиолиты, особенно с высевом P. aeruginosa
4. Двусторонние бронхоэктазы
II. Изменения со стороны желудочно-кишечного тракта
1. Синдром нарушенного кишечного всасывания неясного генеза
2. Мекониевый илеус и его эквиваленты
3. Гиперэхогенность кишечника плода
4. Желтуха обструктивного типа у новорожденных с затяжным течением
5. Цирроз печени
6. Сахарный диабет
7. Гастроэзофагеальный рефлюкс
8. Выпадение прямой кишки
III. Патология со стороны других органов
1. Нарушение роста и развития
2. Задержка полового развития
3. Мужское бесплодие
4. Хронический синусит
5. Полипы носа
6. Синдром псевдо-Барттера
IV. Члены семей больных муковисцидозом
Среди клинических проявлений, характерных для МВ, можно выделить высокоспецифичные и менее специфичные (Табл. 4). Состояния, представленные в левой колонке таблицы, в абсолютном большинстве случаев встречаются у больных МВ. Причиной состояний из правой колонки могут быть другие заболевания, например первичная цилиарная дискинезия, иммунодефицит и т.д.
Таблица 4. Клинические проявления, характерные для МВ

В Таблице 5 представлены особенности проявлений МВ в разные возрастные периоды [1]. Знание этих особенностей помогает специалистам, наблюдающим пациента с теми или иными симптомами, включить МВ в перечень заболеваний для дифференциальной диагностики. Особенно это касается детей раннего возраста, когда клиническая картина еще может быть неполной, но на себя будут обращать внимание некоторые проявления, например мекониевый илеус при рождении или синдром потери солей, не имеющий связи с патологией почек.
Таблица 5. Клинические особенности проявлений МВ в различные возрастные периоды
0-2 года
• Плохая прибавка веса
• Стеаторея
• Рецидивирующие бронхиты/бронхиолиты
• Мекониевый илеус
• Ректальный пролапс
• Гипопротеинемические отеки
• Пневмония/эмпиема
• Синдром псевдо-Барттера
• Затяжная желтуха новорожденных
• Повышенная кровоточивость, связанная с дефицитом витамина K
3-16 лет
• Рецидивирующая инфекция органов дыхания или астма
• Идиопатические бронхоэктазы
• Стеаторея
• Синуситы и назальный полипоз
• Фокальный билиарный цирроз
• Нарушение толерантности к углеводам
• Хроническая интестинальная обструкция, инвагинация
• Тепловой удар с гипонатриемией
Взрослые
• Азооспермия/двусторонняя атрезия семявыносящих протоков
• Бронхоэктазы
• Хронический синусит
• Острый или хронический панкреатит
• Аллергический бронхолегочный аспергиллез
• Фокальный билиарный цирроз
• Портальная гипертензия
• Холелитиаз
• Нарушение толерантности к углеводам
III. Диагностика среди родственников больных
При диагностике МВ должны быть обследованы сибсы больного, независимо от результатов неонатального скрининга. Семье необходимо предложить консультацию генетика для получения информации о типе наследования заболевания и возможностях последующего планирования деторождения (см. Раздел «Генетика муковисцидоза. Молекулярно-генетическая диагностика при муковисцидозе»).
IV. Пренатальная диагностика
• Молекулярно-генетическая диагностика в семьях высокого риска (см. Раздел «Генетика муковисцидоза. Молекулярно-генетическая диагностика при муковисцидозе»).
• Диагноз может быть заподозрен при УЗ-исследовании плода внутриутробно при наличии характерных УЗ-данных гиперэхогенного кишечника [37]. В 50–78% случаев это состояние будет связано с МВ и проявится мекониевым илеусом. Диагноз в этом случае может быть установлен еще до рождения ребенка. В то же время этот признак не является высокоспецифичным для МВ, он может быть транзиторным явлением, а также связанным с другими патологическими состояниями [38]. ДНК-диагностика родителей дает необходимую информацию о наличии мутаций у каждого из родителей и позволяет предполагать заболевание у ребенка при рождении.
V. Преимплантационная диагностика (см. Раздел «Генетика муковисцидоза. Молекулярно-генетическая диагностика при муковисцидозе»)
Основные положения раздела:
1. Нормальный показатель ИРТ, взятый после 8 недель жизни новорожденного, не позволяет исключить МВ.
2. Дети с мекониевым илеусом независимо от уровня ИРТ нуждаются в проведении потовой пробы.
3. Нормальными показателями потовой пробы следует считать в любом возрасте хлориды ≤29 ммоль/л и проводимость пота, эквивалентную <50 ммоль/л хлорида натрия.
4. Необходима тщательная подготовка кожи перед проведением потовой пробы.
5. Ложноположительные результаты неонатального скрининга могут иметь место у носителей мутаций в гене CFTR.
6. Больные МВ – носители «мягких» мутаций в гене CFTR – могут иметь пограничные и отрицательные результаты потовой пробы.
7. Отрицательная потовая проба при положительном неонатальном скрининге и характерной клинической картине требует проведения ДНК-диагностики.
8. Оптимальные сроки установления диагноза и начала наблюдения пациента, выявленного по программе неонатального скрининга, – первые 2 месяца жизни.
9. Обследование и наблюдение новорожденных по программе массового скрининга новорожденных должны проводиться с соблюдением принципов профилактики перекрестного и внутри-больничного инфицирования, оптимально – амбулаторно или в условиях дневного стационара.
Литература
1. Капранов Н.И., Каширская Н.Ю., ред. Муковисцидоз. М.: Медпрактика-М, 2014. 672 с.
2. Шерман В.Д., Капранов Н.И., Каширская Н.Ю., Кондратьева Е.И. Роль неонатального скрининга в оптимизации медицинской помощи больным муковисцидозом в РФ. Медицинская генетика. 2013; 11: 24–29.
3. Bombieri C., Claustres M., De Boeck K., Derichs N., Dodge J., Girodon E., Sermet I., Schwarz M., Tzetis M., Wilschanski M., Bareil C., Bilton D., Castellani C., Cuppens H., Cutting G.R., Drevínek P., Farrell P., Elborn J.S., Jarvi K., Kerem B., Kerem E., Knowles M., Macek M. Jr., Munck A., Radojkovic D., Seia M., Sheppard D.N., Southern K.W., Stuhrmann M., Tullis E., Zielenski J., Pignatti P.F., Ferec C. Recommendations for the classification of diseases as CFTR-related disorders. J. Cyst. Fibros. 2011; 10(2): 86–102.
4. Farrell P.M., Rosenstein B.J., White T.B., Accurso F.J., Castellani C., Cutting G.R., Durie P.R., Legrys V.A., Massie J., Parad R.B., Rock M.J., Campbell P.W. 3rd. Cystic fibrosis foundation. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J. Pediatr. 2008; 153 (2): 4–14.
5. De Boeck K., Wilschanski M., Castellani C., Taylor C., Cuppens H., Dodge J., Sinaasappel M. Cystic fibrosis: terminology and diagnostic algorithms. Thorax. 2006;61:627–635.
6. Smyth А.R., Bell S.C., Bojcin S., Bryon M., Duff A., Flume P., Kashirskaya N., Munck A, Ratjen F., Schwarzenberg S.J., Sermet-Gaudelus I., Southern K.W., Taccetti G., Ullrich G., Wolfe S. European cystic fibrosis society standarts of care working group. Best practice guidelines. J. Cyst. Fibros. 2014; 13 (1): 23-42. https://www.ecfs.eu/ecfs-standards-care/references (дата обращения – 31.12.2016).
7. Dandona P., Hodson M., Bell J., Ramdial L., Beldon I., Batten J. C. Serum immunoreactive trypsin in cystic fibrosis. Thorax. 1981; 36 (1): 60–62.
8. Кусова З.А. Эффективность программы массового обследования новорожденных на муковисцидоз: Автореферат дис. … канд. мед. наук. М., 2011.
9. Castellani C., Southern K.W., Brownlee K., Dankert Roelse J., Duff A., Farrell M., Mehta A., Munck A., Pollitt R., Sermet-Gaudelus I., Wilcken B., Ballmann M., Corbetta C., de Monestrol I., Farrell P., Feilcke M., Férec C., Gartner S., Gaskin K., Hammermann J., Kashirskaya N., Loeber G., Macek M. Jr., Mehta G., Reiman A., Rizzotti P., Sammon A., Sands D., Smyth A., Sommerburg O., Torresani T., Travert G., Vernooij A., Elborn S. European best practice guidelines for cystic fibrosis neonatal screening. J. Cystic Fibrosis. 2009; 8 (3): 153–173.
10. Crossley J.R., Elliott R.B., Smith P.A. Dried-blood spot screening for cystic fibrosis in the newborn. Lancet. 1979; 311(8114): 472–474.
11. Rock M.J., Mischler E.H., Farrell P.M., Wei L.J., Bruns W.T., Hassemer D.J., Laessig R.H. Newborn screening for cystic fibrosis is complicated by age-related decline in immunoreactive trypsinogen levels. Pediatrics. 1990; 85 (6): 1001–1007.
12. Wilcken B., Brown A.R., Urwin R., Brown D.A. Cystic fibrosis screening by dried blood spot trypsin assay: results in 75,000 newborn infants. J. Pediatr. 1983; 102: 383–387.
13. URL: www.newbornbloodspot.screening.nhs.uk (дата обращения – 31.12.2016).
14. Mishra A., Greaves R., Massie J. The Relevance of Sweat Testing for the Diagnosis of Cystic Fibrosis in the Genomic Era. Clin. Biochem. Rev. 2005; 26 (4): 135–153.
15. Gibson L.E., Cooke R.E. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics. 1959; 129: 892–897.
16. Hall E., Lapworth R. Use of sweat conductivity measurements. Annals of Clinical Biochemistry. 2010; 47: 390–392.
17. Sands D., Oltarzewski M., Nowakowska A., Zybert K. Bilateral sweat tests with two different methods as a part of cystic fibrosis newborn screening (CF NBS) protocol and additional quality control. Folia Histochem. Cystobiol. 2010; 30; 48(3): 358–365.
18. Sezer R.G., Aydemir G., Akcan A.B., Paketci C., Karaoglu A., Aydinoz S., Bozaykut A. Nanoduct sweat conductivity measurements in 2664 patients: relationship to age, arterial blood gas, serum electrolyte profiles and clinical diagnosis. J. Clin. Med Res. 2013; 5 (1): 34–41.
19. Langen A.V.,. Dompeling E., Yntema J.B., Arets B., Tiddens H., Loeber G., Dankert-Roelse J. Clinical evaluation of the Nanoduct sweat test system in the diagnosis of cystic fibrosis after newborn screening. Eur J. Pediatr. 2015; 174 (8): 1025–1034.
20. Barben J., Ammann R.A., Metlagel A., Schöni M.H. Conductivity determined by a new sweat analyzer compared with chloride concentrations for the diagnosis of cystic fibrosis. J. Pediatr. 2005; 146: 183–188.
21. Eng W., Le Grys V.A., Shechter M.S., Laughon M.M., Barker P.M. Sweat-testing in pre-term and full-term infants less than 6 weeks of age. Pediatr Pulmonol. 2005; 40: 64–67.
22. Legris V.A., Yankaskas J.R., Quittell L.M., Marshall B.C., Mogayzel P.J. Jr. Diagnostic sweat testing: The Cystic Fibrosis Foundation guidelines. J. Pediatr. 2007; 151(1): 85–89.
23. Farell P.M., Rosenstein B.J., White T.B., Accurso F.J., Castellani C., Cutting G.R., Durie P.R., Legrys V.A., Massie J., Parad R.B., Rock M.J., Campbell P.W. 3rd. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J. Pediatr. 2008; 153(2): 4–14.
24. Knowles M.R., Hohneker K.W., Zhou Z., Olsen J.C., Noah T.L., Ping-Chuanhu, Leigh M.W., Engelhardt J.F., Edwards L.J., Jones K.R., Grossman M., Wilson J.M., Johnson L.G., Boucher R.C. A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N. Engl. J. Med. 1995; 333: 823–831.
25. Derichs N., Sanz J., Von Kanel T., Stolpe C., Zapf A., Tümmler B., Gallati S., Ballmann M. Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: validation and reference data. Thorax. 2010; 65 (7): 594–599.
26. Servidoni M.F., Sousa M., Vinagre A.M., Cardoso S.R., Ribeiro M.A., Meirelles L.R., De Carvalho R.B., Kunzelmann K., Ribeiro A.F., Ribeiro J.D., Amaral M.D. Rectal forceps biopsy procedure in cystic fibrosis: technical aspects and patients perspective for clinical trials feasibility. BMC Gastroenterology. 2013; 20; 13 (1): 91.
27. Webster H.L. Laboratory diagnosis of cystic fibrosis. Crit Rev Clin Lab Sci. 1983: 18 (4): 313–338.
28. Wilschanski M., Zielenski J., Markiewicz D., Tsui L.C., Corey M., Levison H., Durie P.R. Correlation of sweat chloride concentration with classes of the cystic fibrosis transmembrane conductance regulator gene mutations. J. Pediatr. 1995; 127 (5): 705–710.
29. Stewart B., Zabner J., Shuber A.P., Welsh M.J., McCray P.B. Jr. Normal sweat chloride values do not exclude the diagnosis of cystic fibrosis. Am J Respir Crit Care Med. 1995; 151 (3 Pt1): 899–903.
30. Hodson M., Geddes D., Bush A. Cystic fibrosis. Third edition. https://www.amazon.com/Cystic-Fibrosis-Third-Margaret-Hodson/dp/0340907584#reader_0340907584. (дата обращения – 31.12.2016).
31. Guidelines for the performance of the sweat test for the investigation of the CF in the UK. 2014. https:// www.rcpch.ac.uk/child-health/standards-care/clinical-guidelines-and-standards/endorsed-and-supported/respiratory-med#ACB (дата обращения – 31.12.2016).
32. Munck A., Mayell S.J., Winters V. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J. Cyst. Fibros. 2015; 14: 706–713.
33. Gomez L.M., Patuzzo C., Castellani C., Bovo P., Cavallini G., Mastella G., Pignatti P.F. CFTR and cationic trypsinogen mutations in idiopathic pancreatitis and neonatal hypertrypsinemia. Pancreatology. 2001; 1 (5): 538–542.
34. Castellani C., Picci L., Scarpa M. Cystic fibrosis carriers have higher neonatal immunoreactive trypsinogen values than non-carriers. AJMG. 2005; 135A (2): 142–144.
35. Sermet-Gadelous I., Mayell S.J., Southern K.W. Guidelines on the early management of infants diagnosed with cystic fibrosis following newborn screening. J. Cyst. Fibros. 2010; 9 (5): 323–329.
36. Sims E.J., Clark A., McCormick J., Mehta G., Connett G., Mehta A. United Kingdom Cystic Fibrosis Database Steering Committee. Cystic fibrosis diagnosed after 2 months of age leads to worse outcomes and requires more therapy. Pediatrics. 2007; 119: 19–28.
37. Красовский С.А., Петрова Н.В., Степанова А.А., Усачева М.В., Самойленко В.А., Амелина Е.Л., Никонова В.С. Клиническое течение заболевания у взрослых, больных муковисцидозом, – носителей «мягких» мутаций. Пульмонология. 2012; (6): 5–11.
38. De Oronzo M.A. Hyperechogenic fetal bowel: an ultrasonographic marker for adverse fetal and neonatal outcome? J. Prenat. Med. 2011; 5 (1): 9–13.