Синтез гемоглобина что это

Гемоглобин синтезируется в клетках костного мозга. Все необходимые для синтеза гемоглобина составные части поступают с током крови.
Белковая часть молекулы синтезируется как и все простые белки из аминокислот матричным способом.
Синтез гема протекает в несколько стадий под влиянием различных ферментов:
1. Вначале происходит образование дельта-аминолевулиновой кислоты. Это реакция протекает в результате конденсации сукцинил-КоА и глицина в митохондриях под действием фермента аминолевулинатсинтетазы.
2.Следующая реакция протекает в цитоплазме. Происходит образование порфобилиногена в результате реакции конденсации двух молекул дельта-аминолевулиновых кислот.
3.Затем, в результате многоступенчатых реакций из четырех монопиррольных молекул порфобилиногена образуется протопорфирин 1Х, являющийся непосредственным предшественником гема.
4. Протопорфирин IX присоединяет молекулу железа (реакция осуществляется под влиянием фермента гемсинтетазы или феррохелатазы) и образуется гем, который затем используется для биосинтеза всех гемопротеидов. Оба фермента, участвующие в синтезе ПБГ, регулируемые, они ингибируются гемом и НЬ. Поэтому гем не образуется в избытке или недостатке. Также строго в определенном количестве образуется и белковая часть Нb, т. к. ее синтез может происходить только в присутствии тема, и образующиеся полипептидные цепи тут же соединяются с гемом. При низкой концентрации гема, когда нарушается его синтез, образование гемоглобина также замедляется.
Каждая из образовавшихся полипетидных цепей глобина присоединяются кодному гему, образуя моном ер гемоглобиан. 4 таких мномера, объединивщись, образуют гемоглобин.
Основной функцией гемоглобина является перенос кислорода из легких к тканям и перенос углекислого газа от тканей к легким, участие в поддержке рН крови. Свои функции гемоглобин выполняет только в составе эритроцита. Продолжительность жизни эритроцита 110-120 дней. Затем эритроцит подвергается гемолизу
3. Распад гемоглобина. Превращение билирубина в желудочно- кишечном тракте. Свободный и связанный билирубин. Свойства.
При гемолизе эритроцитов гемоглобин попадает в кровь и соединяется с белком гаптоглобином, в виде комплекса гемоглобин-гаптоглобин (Нр-Нb) транспортируется в клетки макрофагально-моноцитарной системы (ММС): это Купферовы клетки печени, клетки лимфоузлов, селезенки, пейеровых бляшек в кишечнике.
Процесс начинается с окислительного расщепления метинового мостикамежду первым и вторым пиррольными кольцами и образуется вердоглобин. Затем от вердоглобина отщепляется глобин, железо и образуется биливердин (зеленого цвета), вещество линейной структуры. Железо соединяется с b-глобулинами и в виде трансферина доставляется в печень и селезенку, где депонируется в виде ферритина. Глобин распадается так же как и все простые белки до аминокислот.
Биливердин восстанавливается за счет НАДФН2 в неконьюгированный,
свободный билирубин, который не растворим в воде и является токсичным соединением. Свободный билирубин выходит из клеток ММС, соединяется с
альбуминами и поступает в гепатоциты. В крови он называется непрямым потому, что дает реакцию с реактивом Эрлиха не сразу, а после добавления в сыворотку крови кофеинового реактива или спирта для осаждения белка.
В Купферовых клетках печени распад гемоглобина также начинается с
образования вердоглобина, затем биливердина. В печени непрямой билирубин обезвреживается в гепатоцитах путем реакции конъюгации, соединяясь с одной или двумя молекулами глюкуроновой кислоты, образуя моно- или диглюкуронид билирубина. Такой билирубин называется конъюгированным и
связанным и прямым. Этот билирубин хорошо растовряется в воде, не обладает токсическими свойствами. Биливердин и прямой билирубин собираются в желчном пузыре, придавая желчи оливковый цвет и потому их относят к пигментам желчи. Желчь поступает в тонкий кишечник, но в желчном протоке прямой билирубин, теряя глюкуроновые кислоты, снова превращается в непрямой. Биливердин проходит через весь кишечник не изменяя своей химической структуры и удаляется с калом, окрашивая его в зеленоватый цвет, т.е. он является пигментом кала. А непрямой билирубин в кишечнике восстанавливается до мезобилиногена (уробилиногена), часть которого всасывается в воротную вену и возвращается в печень, где распадается до бесцветных моно- и дипирролов. Последние выводятся через почки вместе с мочой.
Большая часть мезобилиногена поступает в толстый кишечник, где под
влиянием ферментов микроорганизмов восстанавливается в стеркобилиноген. Часть стеркобилиногена, всасываясь в кровь через геморроидальные вены, попадает в почки. В моче под действием света и воздуха происходит окисление стеркобилиногена до стеркобилина, который придает моче желтый цвет, т.е. является пигментом мочи. Остальная часть стеркобилиногена окисляется в толстом кишечнике на свету до стеркобилина и вместе с биливердином является пигментом кала, придавая ему коричнево-зеленый цвет.
У грудных детей в кишечнике нет гнилостных бактерий, поэтому
билирубин не превращается в стеркобилиноген и выводится как таковой. Соответственно цвет кала у детей обусловлен биливердином и билирубином (желто-зеленый).
У детей в первые три месяца эмбрионального периода образуется эмбриональный гемоглобин. Затем он преобразуется в фетальный (гемоглобин F), который доминирует вплоть до рождения ребенка. После рождения в течение первого месяца жизни фетальный гемоглобин постепенно заменяется на гемоглобин взрослого (гемоглобин А), отличающегося составом полипептидных цепей. Эмбриональный и фетальный гемоглобин обладают более высоким сродством к кислороду по сравнению с гемоглобином взрослого.
Пигменты желчи, кала и мочи.
При распаде гемоглобина образуются пигменты желчи, кала и мочи.
Пигменты желчи: биливердин (зеленого цвета), связанный билирубин (глюкурониды билирубина –желтого цвета). Цвет желчи зависит от соотношения этих пигментов.
Пигменты кала: биливердин (зеленого цвета), стеркобилин (коричневого цвета)
Пигмент мочи: стеркобилин
Цвет сыворотки крови тоже зависит в определенной степени от наличия в ней билирубина. В норме количество общего билирубина в крови равно 8—20 мкмоль/л, на долю непрямого билирубина приходится 75- 100%, а прямого от 0 до 25%. Количество прямого билирубина незначительно. Прямой билирубин проходит через пачечную ткань, и появляется в моче, непрямой билирубин в моче появиться не может, вследствие его нерастворимости в воде.
Гем является небелковой частью многих гемопротеинов:
- гемоглобин (до 85% общего количества гема организма), локализованный в эритроцитах и клетках костного мозга,
- миоглобин скелетных мышц и миокарда (до 17%),
- цитохромы дыхательной цепи,
- ферменты цитохромоксидаза, цитохром P450, гомогентизатоксидаза, миелопероксидаза, каталаза и глутатионпероксидаза, тиреопероксидаза и т.д. – менее 1%.
Строение и синтез гема
Гем – структура, включающая в себя порфириновое кольцо (состоящее из 4 пиррольных колец) и иона Fe2+. Железо связывается с порфириновым кольцом двумя координационными и двумя ковалентными связями.
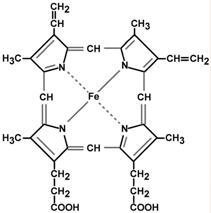
Строение гема
Синтез гема в основном идет в предшественниках эритроцитов, в клетках печени, почек, слизистой кишечника и в остальных тканях. Первая реакция синтеза с участием δ-аминолевулинат-синтазы (греч. δ – “дельта”) происходит в митохондриях. Следующая реакция при участии аминолевулинатдегидратазы (порфобилиноген-синтазы) протекает в цитозоле, здесь из двух молекул δ‑аминолевулиновой кислоты образуется циклический порфобилиноген (монопиррол).
Синтез порфобилиногена
После синтеза порфобилиногена четыре его молекулы конденсируются в гидроксиметилбилан, который далее превращается в уропорфириноген типа I и уропорфириноген типа III. В синтезе обоих видов порфиринов принимает участие уропорфириноген I-синтаза, в образовании уропорфириногена III дополнительно принимает участие фермент уропорфириноген III-косинтаза.
Судьба обоих типов уропорфириногена двояка: они могут окисляться до уропорфирина (на рисунке не показано) или декарбоксилироваться до копропорфириногена соответствующего типа.
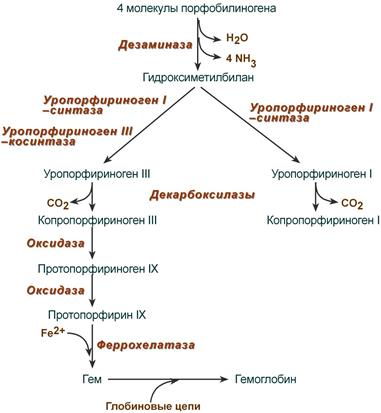
Синтез гема из порфобилиногена
Копропорфириноген III возвращается в митохондрии и окисляется в протопорфириноген IX и далее в протопорфирин IX. Последний после связывания с железом образует гем, реакцию катализирует феррохелатаза (гемсинтаза).
Скорость синтеза глобиновых цепей зависит от наличия гема, он ускоряет биосинтез “своих” белков.
Названия пигментов (уропорфирины и копропорфирины) были даны веществам по источнику их первоначального выделения, при этом восстановленные бесцветные формы называют порфириногенами. Для порфиринов характерно наличие изомерии вследствие различного расположения радикалов, что нашло отражение в порядковых номерах изомеров.
Регуляция синтеза гема
Основным регуляторным ферментом синтеза гема является аминолевулинатсинтаза.
1. Гем :
- напрямую оказывает отрицательный аллостерический эффект на фермент,
- влияет на транскрипцию фермента. После взаимодействия с молекулой белка-репрессора формирует активный репрессорный комплекс, связывается с ДНК и подавляет транскрипцию, мРНК для фермента не образуется и синтез фермента прекращается.
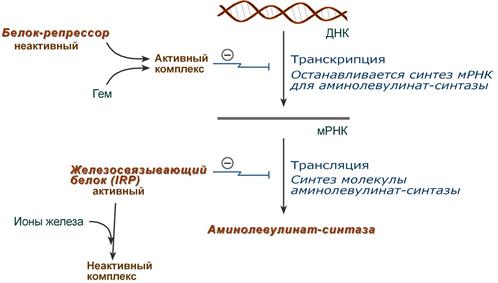
Регуляция синтеза аминолевулинатсинтазы
2. Ионы железа. Их достаточное количество оказывает положительный эффект при синтезе молекулы аминолевулинатсинтазы.
В клетке имеется железосвязывающий белок (англ. IRP, iron-responsive element-binding proteins – белок, связывающий железочувствительный элемент), который в отсутствии ионов железа обладает сродством к железочувствительному участку IRE (англ. iron-responsive element ) на матричной РНК фермента. Это связывание блокирует трансляцию мРНК в рибосоме, т.е. подавляет синтез белковой цепи.
При наличии ионов железа они связываются с железосвязывающим белком, образуя с ним неактивный комплекс, и это инициирует синтез фермента.
3. Положительным модулятором аминолевулинатсинтазы служит внутриклеточная гипоксия, которая в эритропоэтических тканях индуцирует синтез фермента.
4. В печени повышению активности аминолевулинатсинтазы способствуют различные соединения, усиливающие работу микросомальной системы окисления (жирорастворимые вещества, стероиды) – при этом возрастает потребление гема для образования цитохрома Р450, и снижается внутриклеточная концентрация свободного гема. В результате происходит усиление синтеза фермента.
Синтезированный в митохондриях гем
индуцируется синтез цепей глобина на
полирибосомах. Гены цепей глобина
расположены в 11 и 16 хромосоме.
Цепи глобина формируют глобулы и
соединяются с гемом. 4 глобулы нековалентно
соединяются в гемоглобин.
Гемоглобин начинает синтезироваться
на стадии базофильного эритробласта,
а заканчивается у ретикулоцитов. В
ретикулоцитах также идет синтез пуринов,
пиримидинов, фосфатидов, липида.
Чувствительным биохимическим индикатором
для отличия ретикулоцитов от зрелых
клеток является утрата последними
глутаминазы. Глутамин в ретикулоцитах
– источник углерода для синтеза порфирина
и азота для синтеза пурина.
Строение гемоглобина
Гемоглобин– тетрамерный
хромопротеин, имеет массу 64,5кДа, состоит
из 4 гемов и 4 глобинов. Глобины представлены
полипептидными цепями различных типов,,,и т.д.-цепь содержит
141 АК, а- цепь – 146
АК. Отдельные участки полипептидных
цепей образуют правозакрученные-спирали, особое
расположение в пространстве которых
формирует глобулы. Глобула-субъединицы
содержит 8-спиралей,
а-субъединицы –7.
Гем располагается в щелях между Е иFспиралями глобина, прикрепляясь через
гистидинF8 к спиралиFс помощью 5 координационной
связи железа. Гидрофобные остатки
аминокислот окружающие гем, препятствуют
окислению железа водой. 4 глобулы с
участием гидрофобных, ионных и водородных
связей формируют шарообразный тетрамер
гемоглобина. Максимально прочные связи,
в основном за счет гидрофобных связей,
образуются между-
и-глобулами. В
результате образуются 2 димера11и22.
Димеры соединяются между собой в основном
полярными (ионными и водородными)
связями, поэтому взаимодействие димеров
зависит от рН. Димеры легко перемещаются
друг относительно друга. В центре
тетрамера глобулы прилегают друг к
другу неплотно, образуя полость.
Функции гемоглобина
Обеспечивают
перенос кислорода от легких к тканям.
В сутки около 600литров;Участвует
в переносе углекислого газа и протонов
от тканей к легким;Образует
гемоглобиновый буфер, регулирует КОС
крови.
Производные гемоглобина
Гемоглобин
со свободной шестой координационной
связью железа в составе гема называется
апогемоглобином.
Шестая
координационная связь может связывать
различные лиганды, с образованием
следующих производных гемоглобина:
оксигемоглобинHbО2(Fe2+)
– соединение молекулярного кислорода
с гемоглобином. Процесс называется
оксигенацией; обратный процесс –
дезоксигенацией.карбоксигемоглобинHbСО (Fe2+).
Связь гема с СО в двести раз прочнее,
чем с О2. В норме в крови содержится
1%HbСО. У курильщиков к
вечеру концентрацияHbСО
достигает 20%. При отравлении СО, из-за
недостаточного снабжения тканей
кислородом может наступить смерть.метгемоглобинHbОН (Fe3+).
Образуется при воздействии на гемоглобин
окислителей (оксидов азота, метиленового
синего, хлоратов). В норме в крови
содержится<1%HbОН.
Накопление метгемоглобина при некоторых
заболеваниях (например, нарушение
синтеза ГЛ-6-фосфатДГ), отравлении
окислителями может стать причиной
смерти, так как метгемоглобин не способен
к переносу кислорода;цианметгемоглобинHbСN(Fe3+).
Образуется при присоединении СN-к метгемоглобину. Эта реакция спасает
организм от смертельного действия
цианидов. Поэтому для лечения отравлений
цианидами применяют метгемоглобинообразователи
(нитритNa);
Карбгемоглобинобразуется,
когда гемоглобин связывается с СО2.
Однако СО2присоединяется не к
гему, а кNН2– группам
глобина, с образованием карбаматов:
HbNH2 +CO2=HbNHCOO-+H+
Карбгемоглобин выводит из организма
10-15% СО2.
ДезоксигемоглобинHb(Fe2+). Форма гемоглобина
не связанная скислородом.
Дезоксигемоглобин связывает больше
СО2, чем оксигемоглобин.
В
цитохромахгем присоединяется к
белковой части через 5 и 6 координационные
связи железа (через гистидин и метионин
Е иFспиралей). Занятость
всех координационных связей не позволяет
цитохромам присоединять лиганды, поэтому
они могут переносить только по 1 электрону.
Механизм насыщения гемоглобина
кислородом
Гемоглобин присоединяет О2последовательно, по одной молекуле на
каждый гем.
В апогемоглобине,благодаря
координационной связи с белковой частью,
атом железа выступает из плоскости гема
в направлении гистидинаF8.
Присоединение О2к шестой
координационной связи железа вызывает
его перемещение в плоскость гема, за
ним перемещаются гистидинF8
и полипептидная цепь, в состав которой
он входит.
Происходит изменение конформации
текущего протомера и связанных с ним
оставшихся протомеров. При этом у
протомеров возрастает сродство к
кислороду, в результате каждый следующий
кислород присоединяется к гемоглобину
лучше предыдущего. Четвертая молекула
кислорода присоединяется к гемоглобину
в 300 раз легче, чем первая молекула.
Обратный процесс аналогичен, чем больше
О2отдают протомеры, тем легче
идет отщепление последующих молекул
О2.
Кривая диссоциации кислорода для
гемоглобина
Кооперативность в работе протомеров
гемоглобина формирует сигмовидный
характер кривой насыщения его кислородом
в зависимости от парциального давления
кислорода.
S–образная кривая насыщения
гемоглобина кислородом имеет важное
биологическое значение.
Во-первых, пологий участокS–образной
кривой (выше 60мм.рт.ст.)
обеспечивает максимальное насыщение
гемоглобина кислородом в легких, даже
если концентрация кислорода в альвеолярном
воздухе заметно снижена. Например, в
альвеолярной крови приРО2=95
мм.рт.ст. гемоглобин насыщается кислородом
на 97%, а при РО2=60
мм.рт.ст. – на 90%.
Во-вторых,Крутой наклон
среднего участка S–образной
кривой (от 10 до40 мм.рт.ст.)
обеспечивает максимальный переход
кислорода от гемоглобина к тканям.
В области венозного конца
капилляра приРО2
= 40 мм.рт.ст. гемоглобин насыщен кислородом
на 73%. При снижении РО2
на 5 мм.рт.ст. насыщение гемоглобина
кислородом уменьшается на 7%.
Аллостерическая регуляция насыщения
гемоглобина кислородом
Кроме РО2
на насыщение гемоглобина
кислородом влияют и другие факторы,
например, рН, температура, давление,
концентрация 2,3-ДФГ,РСО2.
Увеличение температуры, присоединение
к гемоглобину Н+, 2,3-ДФГ, СО2уменьшает сродство гемоглобина к
кислороду, при этом кривая диссоциации
оксигемоглобина сдвигается вправо и
гемоглобин легче отдает кислород тканям.
Эффект Бора
Влияние рН на характер кривой диссоциации
оксигемоглобина называется эффектом
Бора(по имени датского физиолога
Христиана Бора, впервые открывшего этот
эффект).
Гемоглобин в дезоксигенерированном
состоянии имеет более высокое сродство
к протонам, чем оксигемоглобин. Другими
словами R – форма (оксигенерированная)
является более сильной кислотой, чем
Т-форма (дезоксигенерированная). Поэтому
когда дезоксигемоглобин в легких
присоединяет кислород, происходит
переход в R – форму и разрыв некоторых
связей, в результате чего и высвобождаются
протоны, ответственные за эффект Бора.
Наоборот, при высвобождении кислорода
образуется Т-структура и разорванные
связи между субъединицами должны быть
восстановлены, и протоны вновь
присоединяются к остаткам гистидина
в - цепях. Таким
образом, протонирование гемоглобина
снижает его сродство к О2 и увеличивает
потребление О2 в ткани.
Эффект Бора имеет важное физиологическое
значение. Образующийся в тканях СО2
должен транспортироваться в легкие. Он
поступает в эритроциты по градиенту
напряжения. В них фермент карбоангидраза
превращает его в Н2СО3, который диссоциирует
на бикарбонат, ион и протон. Последний
сдвигает равновесие влево в уравнении
(1).
Hb + 4 O2= Hb (О2)4 + (H+)n
Где n – величина порядка 2; число зависит
от целого комплекса параметров, тем
самым заставляя Hb О2 отдавать свой
кислород.
НСО3- пассивно продвигается через ионный
канал по градиенту концентрации в
сыворотку.
Продвижение НСО3- не сопровождается
перемещением Н+, поскольку нет канала,
позволяющего ему пройти через мембрану
эритроцитов. Для сохранения ионного
равновесия при выходе НСО3- из клетки,
Cl- перемещаются внутрь её через тот же
ионный канал. Такое двойное перемещение
известно как хлоридный сдвиг (сдвиг
Хамбургера).
Растворенный НСО3- движется вместе с
венозной кровью обратно в легкие. Здесь
высвобождение протона из гемоглобина
при оксигениции приводит к образованию
НСО3- (по принципу Ле-Шателье).
НСО3-+ Н+= Н2СО3-,
что позволяет карбоангидразе образовать
СО2.
Разрушение НСО3- в эритроците обуславливает
вхождение в него НСО3- из сыворотки, так
что в легких происходит обратный
хлоридный сдвиг, приводящий к выведению
СО2 с выдыхаемым воздухом.
Аллостерическая регуляция сродства
гемоглобина к кислороду 2,3-ДФГ
2,3-ДФГ снижает сродство гемоглобина к
кислороду и, таким образом, повышает
отдачу кислорода тканям. Если кровь
израсходовала весь свой запас ДФГ,
гемоглобин остается фактически насыщенным
кислородом. При акклиматизации в условиях
высокогорья содержание ДФГ в эритроцитах
резко увеличивается. ДФГ является
аллотерическим лигандом, так как
связывается с гемоглобином в другом по
сравнению с О2 участком. ДФГ встраивается
в полость тетрамерной молекулы
гемоглобина, полость образована остатками
всех 4 протомеров.
В Т – форме (дезоксигенерированной)
молекулы Hb имеются дополнительные
связи, и поэтому размер центральной
полости больше, чем в R – форме
(дезоксигемоглобине). Поэтому ДФГ
взаимодействует только с Т – формой
стабилизируя её, путем образования
связи между атомами кислорода ДФГ и
тремя положительно заряженными группами
в каждой из - цепей.
В легких при высоком парциальном давлении
кислород взаимодействует с Hb, изменяется
конформация белка, уменьшается центральная
полость и ДФГ вытесняется из гемоглобина.
Виды гемоглобинов
Гемоглобины различаются по белковой
части. Бывают физиологические и аномальные
виды гемоглобинов. Физиологические
образуются на разных этапах нормального
развития организма, а аномальные –
вследствие нарушения последовательности
аминокислот в глобине физиологических
видов гемоглобина.
Физиологические виды гемоглобина
1) эмбриональные гемоглобины(Gover I, Gover II). На ранних этапах развития
плода в первые недели развития, когда
в желточном мешке возникают очаги
кроветворения начинается синтез-цепей
(эпсилон). Из четырёхцепей образуется гемоглобин Gover I. Затем
у эмбриона, длина которого не превышает
2,5см, начинается синтез-цепей,
которые вместе-цепями
образуют гемоглобин Gover II (22). Затем синтез-цепей прекращается
и Gover гемоглобины полностью исчезают у
трехмесячного эмбриона. Если они остаются
у новорожденного, то это признак
врожденной аномалии развития.
2) фетальный гемоглобин– HbF (от
латинского fetus – плод). Фетальный
гемоглобин сменяет эмбриональные
гемоглобины, вместо эпсилон – цепей (- цепей) начинают синтезироваться
гамма-цепи (- цепи).
HbF состоит из 2и 2цепей. HbF – является главным гемоглобином
плода и составляет к моменту рождения
50-80% всего гемоглобина. HbF имеет более
высокое сродство к кислороду, что
позволяет ему забирать кислород от
гемоглобина матери и передавать его
тканям плода. Эта особенность связана
с низким сродством HbF к 2,3-ФГК.
Кроме перечисленных основных видов
гемоглобинов плода, у здорового плода
выделяются и другие виды гемоглобинов:
например, гемоглобин Bart`s, (4),
Portland–1 (S22).
Схема
электрофореза
гемоглобина
здорового плода
+
А1
F
А2
Gower I








Bart`s
Portland
-1
Gower II
3) гемоглобин А1–
тетрамер (22)
составляет около 98% гемоглобина
эритроцитов взрослого человека. Начинает
синтезироваться на 8 месяце развития
плода.
4) гемоглобин А2–
тетрамер (22).
Его содержание в эритроцитах взрослого
человека равно 2%. Гемоглобин А2,
также как и гемоглобин F, обладает более
высоким сродством к кислороду по
сравнению с гемоглобином А1.
5) гемоглобин А3(22)
образуется по мере старения эритроцита,
при присоединении к цистеину-цепи
глутатиона.
6) гемоглобин А1С– гликозилированный гемоглобин А.
Аномальные виды гемоглобинов
Аномальные гемоглобины возникают в
результате мутации генов, кодирующих
ицепи. Известно несколько сотен мутантных
гемоглобинов человека (в большинстве
случаев функционально активных).
Таблица №1 замена аминокислот в ипептидных цепях
гемоглобина
тип гемоглобина | нормальный | замена |
С | глу 6 в - цепи | лиз |
Д | лей 28 в - цепи | глу |
Е | глу 26 в - цепи | лиз |
G | глу 43 в - цепи | ала |
GpH | асл 68 в - цепи | лиз |
J | лиз 16 в - цепи | асл |
М | вал 67 в - цепи | глу |
О | глу 116 в - цепи | лиз |
S | глу 6 в - цепи | вал |
Болезни гемоглобинов
Болезни гемоглобинов называют
гемоглобинозами, их насчитывают
более 200.
Гемоглобинозы делятся на гемоглобинопатии
и таласемии.
Гемоглобинопатии, возникают в
результате точечных мутаций в структурных
генах, кодирующих полипептидные цепи
гемоглобина. Поэтому в крови появляется
аномальный гемоглобин.
Серповидноклеточная анемия–
классический пример наследственной
гемоглобинопатии. В норме в-субъединицах
гемоглобина в шестом положении находится
гидрофильная глутаминовая кислота. В
гемоглобине S глутаминовая кислота
заменена на гидрофобный валин. Такая
замена приводит к появлению на поверхности-субъединицы
гидрофобного («липкого») участка, который
соединяется с гидрофобным карманом
другой молекулы гемоглобина S. Происходит
полимеризация гемоглобина S и его
осаждение в виде длинных волокон. Длинная
волокнистая структура нарушает нормальную
форму эритроцитов, превращая её из
двояковогнутого диска в серповидную,
которая имеет тенденцию блокировать
капилляры. Такие эритроциты преждевременно
разрушаются, способствуя развитию
анемии. Если поражены обе гомологичные
хромосомы, заболевание может оказаться
смертельным. Заболевание широко
распространено в географических зонах,
где наиболее часто встречается
злокачественная форма малярии. Высокий
показатель заболеваемости можно
объяснить положительной селекцией
генома носителей аномальных генов.
Серповидная красная кровяная клетка
«неудобна» для развития малярийного
плазмодия.
Существенное ухудшение состояния
больных наблюдается в условиях высокогорья
при низких давлениях кислорода. Это
связано с тем, что полимеризоваться
способна только дезоксиформа S гемоглобина.
Так как в молекуле оксиформы S-гемоглобина
нет гидрофобного кармана («липкого
участка»), и она не способна к полимеризации.
Талассемия– генетическое заболевание,
обусловленное отсутствием или снижением
синтеза одной из цепей гемоглобина. При
данном заболевании отсутствуют дефекты
в структурных генах, кодирующих,,,-цепи.
Причиной талассемий являются мутации
генов-операторов, контролирующих
транскрипцию структурных генов ,,,-цепей гемоглобина.
В результате несбалансированного
образования глобиновых цепей образуются
тетрамеры гемоглобина, состоящие из
одинаковых протомеров.
В зависимости от того, формирование
какой глобиновой цепи нарушается,
выделяют ,
, ,
–
талассемии.
Талассемии делятся так же на гомозиготныеигетерозиготные.
Гомозиготная -талассемия– формирование-цепи
полностью подавляется. Симптомы
заболевания появляются приблизительно
через полгода после рождения, когда
происходит полное переключение синтеза-цепи гемоглобина
F на-цепь. У ребенка
прогрессирует анемия. Увеличиваются
селезенка и печень. Лицо приобретает
монголоидные черты (из-за чрезмерного
разрастания костного мозга скулы
выдаются вперед, нос приплюснут), при
рентгенологическом исследовании черепа
наблюдается феномен «игл ежа» («hair –
standing –on –end»). В попытке восполнить
эритроциты, утраченные в результате не
эффективного эритропоэза и увеличении
гемолиза, ткани черепа, чрезмерно
разрастаясь и гипертрофируясь, порождают
такое изменение медуллярной пластинки.
α-талассемия
– недостаток образования
α-глобиновых цепей приводит к нарушению
образования HbF
у плода. Избыточные
γ-цепи образуют тетрамеры, называемые
гемоглобином
Барта. Этот гемоглобин при физиологических
условиях имеет повышенное сродство
к кислороду и не проявляет кооперативных
взаимодействий между протомерами. В
результате гемоглобин Барта не
обеспечивает
развивающийся плод необходимым
количеством
кислорода, что приводит к тяжёлой
гипоксии. При α-талассемии
отмечают высокий процент внутриутробной
гибели плода.
Выжившие новорождённые при переключении
с γ- на β-ген синтезируют β-тетрамеры
или
НbН,
который, подобно гемоглобину Барта,
имеет слишком высокое сродство к
кислороду,
менее стабилен, чем НbА
и быстро разрушается.
Это ведёт к развитию у больных тканевой
гипоксии и к смерти вскоре после рождения.
Для всех этих заболеваний характерны
некоторые общие закономерности:
1). нарушаются пропорции в составе
гемоглобина крови. Например, при -
талассемии в крови появляется 15%
гемоглобина А2, 15 – 60% гемоглобина
F;
2). эритроциты приобретают не нормальную
форму (мишеневидную, каплевидную). Такие
эритроциты в пределах 1 дня захватываются
ретикулярной соединительной тканью
(например, селезенкой) и подвергаются
распаду (по этой причине селезёнка
оказывается гипертрофированной), что
приводит к развитию гемолитической
анемии.
Соседние файлы в папке 7 модуль
- #
- #
- #
- #
- #
- #
- #