Сродством к гемоглобину обладает

Оглавление темы “Вентиляция легких. Перфузия легких кровью.”: Сродство гемоглобина к кислороду. Изменение сродства гемоглобина к кислороду. Эффект Бора.Молекула гемоглобина может находиться в двух формах — напряженной и расслабленной. Расслабленная форма гемоглобина имеет свойство насыщаться кислородом в 70 раз быстрее, чем напряженная. Изменение фракций напряженной и расслабленной формы в общем количестве гемоглобина в крови обусловливает S-образный вид кривой диссоциации оксигемоглобина, а следовательно, так называемое сродство гемоглобина к кислороду. Если вероятность перехода от напряженной формы гемоглобина к расслабленной больше, то возрастает сродство гемоглобина к кислороду, и наоборот. Вероятность образования указанных фракций гемоглобина изменяется в большую или меньшую сторону под влиянием нескольких факторов. Основной фактор — это связывание кислорода с геминовой фуппой молекулы гемоглобина. При этом чем больше геминовых фупп гемоглобина связывают кислород в эритроцитах, тем более легким становится переход молекулы гемоглобина к расслабленной форме и тем выше их сродство к кислороду. Поэтому при низком Р02, что имеет место в метаболически активных тканях, сродство гемоглобина к кислороду ниже, а при высоком Р02 — выше. Как только гемоглобин захватывает кислород, повышается его сродство к кислороду и молекула гемоглобина становится насыщенной при связывании с четырьмя молекулами кислорода.
Когда эритроциты, содержащие гемоглобин, достигают тканей, то кислород из эритроцитов диффундирует в клетки. В мышцах он поступает в своеобразного депо кислорода — в молекулы миоглобина, из которого кислород используется в биологическом окислении мышц. Диффузия кислорода из гемоглобина эритроцитов в ткани обусловлена низким Р02 в тканях — 35 мм рт. ст. Внутри клеток тканей напряжение кислорода, необходимое для поддержания нормального метаболизма, составляет еще меньшую величину — не более 1 кПа. Поэтому кислород путем диффузии из капилляров достигает метаболически активных клеток. Некоторые ткани приспособлены к низкому содержанию Р02 в капиллярах крови, что компенсируется высокой плотностью капилляров на единицу объема тканей. Например, в скелетной и сердечной мышцах Р02 в капиллярах может снизиться чрезвычайно быстро во время сокращения. В мышечных клетках содержится белок миоглобин, который имеет более высокое сродство к кислороду, чем гемоглобин. Миоглобин интенсивно насыщается кислородом и способствует его диффузии из крови в скелетную и сердечную мышцы, где он обусловливает процессы биологического окисления. Эти ткани способны экстрагировать до 70 % кислорода из крови, проходящей через них, что обусловлено снижением сродства гемоглобина к кислороду под влиянием температуры тканей и рН. Эффект рН и температуры на сродство гемоглобина к кислороду. Молекулы гемоглобина способны реагировать с ионами водорода, в результате этой реакции происходит снижение сродства гемоглобина к кислороду. При насыщении гемоглобина менее 100 % низкое рН понижает связывание кислорода с гемоглобином — кривая диссоциации оксигемоглобина смещается вправо по оси х. Это изменение свойства гемоглобина под влиянием ионов водорода называется эффектом Бора. Метаболически активные ткани продуцируют кислоты, такую как молочная, и С02. Если рН плазмы крови снижается от 7,4 в норме до 7,2, что имеет место при сокращении мыщц, то концентрация кислорода в ней будет возрастать вследствие эффекта Бора. Например, при постоянном рН 7,4 кровь отдавала бы порядка 45 % кислорода, т. е. насыщение гемоглобина кислородом снижалось до 55 %. Однако когда рН снижается до 7,2, кривая диссоциации смещается по оси х вправо. В результате насыщение гемоглобина кислородом падает до 40 %, т. е. кровь может отдавать в тканях до 60 % кислорода, что на 1/з больше, чем при постоянном рН.
Метаболически активные ткани повышают продукцию тепла. Повышение температуры тканей при физической работе изменяет соотношение фракций гемоглобина в эритроцитах и вызывает смещение кривой диссоциации оксигемоглобина вправо вдоль оси х. В результате большее количество кислорода будет освобождаться из гемоглобина эритроцитов и поступать в ткани. Эффект 2,3-дифосфоглицерата (2,3-ДФГ) на сродство гемоглобина к кислороду. При некоторых физиологических состояниях, например при понижении Р02 в крови ниже нормы (гипоксия) в результате пребывания человека на большой высоте над уровнем моря, снабжение тканей кислородом становится недостаточным. При гипоксии может понижаться сродство гемоглобина к кислороду вследствие увеличения содержания в эритроцитах 2,3-ДФГ. В отличие от эффекта Бора, уменьшение сродства гемоглобина к кислороду под влиянием 2,3-ДФГ не является обратимым в капиллярах легких. Однако при движении крови через капилляры легких эффект 2,3-ДФГ на снижение образования оксигемоглобина в эритроцитах (плоская часть кривой диссоциации оксигемоглобина) выражен в меньшей степени, чем отдача кислорода под влиянием 2,3-ДФГ в тканях (наклонная часть кривой), что обусловливает нормальное кислородное снабжение тканей. – Также рекомендуем “Углекислый газ. Транспорт углекислого газа.” |
Варианты гемоглобина в онтогенезе
Производные гемоглобина
Гемоглобин человека
Гемоглобин – сложный железосодержащий белок, относится к классу гемопротеинов. Выполняет две важные функции:
· перенос кислорода из легких к периферическим тканям;
· участие в переносе СО2 и протонов из периферических тканей в легкие.
Молекула гемоглобина взаимодействует с различными лигандами, образуя производные гемоглобина.
1. Дезоксигемоглобин – ННb – не связанный с кислородом и содержащий гем с двухвалетным железом Fe2+.
2. Оксигемоглобин– ННbO2 – полностью оксигенированный гемоглобин, связанный с четырьмя молекулами кислорода.
3. Карбгемоглобин – ННbCO2 – гемоглобин, связанный с СО2. Выполняет функцию выведения СО2 из тканей к легким. Соединение нестойкое, легко диссоциирует в легочных капиллярах. Этим путем выводится до 10-15% СО2.
4. Карбоксигемоглобин– ННbСО – образуется при отравлении оксидом углерода (II). Сродство гемоглобина к СО примерно в 300 раз выше, чем к кислороду, при этом гемоглобин теряет способность связывать кислород и наступает смерть от удушья.
5. Метгемоглобин– MetHb – образуется при действии окислителей (нитрит натрия, нитробензол). Содержит железо в трехвалентной форме Fe3+ и теряет способность к переносу кислорода. В норме образуется небольшое количество метгемоглобина – примерно 0,5 % в сутки.
Количество и состав фракций гемоглобина изменяется в процессе онтогенеза. Все гемоглобины представляют собой тетрамеры, построенные из разного набора субъединиц (α, β, γ, δ) и преимущественно образуются на разных этапах развития организма человека – от эмбрионального до взрослого состояния. Различают следующие физиологические типы гемоглобинов: примитивный гемоглобин НbР, фетальный гемоглобин HbF (fetus – плод), гемоглобин взрослых HbA, HbA2, HbA3 (adultus – взрослый).
Примитивный гемоглобин– синтезируется в эмбриональном желточном мешке через несколько недель после оплодотворения. Состоит из двух α- и двух ε-цепей (2α, 2ε). Через две недели после формирования печени плода в ней начинает синтезироваться HbF, который к шести месяцам полностью замещает НbР.
Фетальный гемоглобин – синтезируется в печени и костном мозге плода до периода его рождения. Состоит из двух α- и двух γ-цепей (2α, 2γ). Характеризуется более высоким сродством к кислороду и обеспечивает эффективную доставку кислорода к эмбриону из системы кровообращения матери. HbF является главным типом гемоглобина плода. Кровь новорожденного содержит до 80% HbF, но к концу 1-го года жизни он почти целиком заменяется на HbA. В крови взрослого человека присутствует в минимальном количестве – до 1,5% от общего количества гемоглобина.
Гемоглобин А – основной гемоглобин взрослого человека (96 % от общего количества). Начинает синтезироваться в клетках костного мозга уже на 8-м месяце развития плода. HbA состоит из двух α- и двух β-цепей.
Минорные гемоглобины:
1) HbA2 – 2α 2δ, в крови взрослого человека примерно 2,6 % HbA2. Обладает большим сродством к кислороду.
2) HbA3 – 2α 2β, однако имеются изменения в строении β-цепей по сравнению с HbA. Появляется в крови в небольших количествах при старении.
Все структурные аномалии белковой части гемоглобина называют гемоглобинозами. Различают:
· гемоглобинопатии;
· талассемии.
Гемоглобинопатии – наследственные изменения структуры какой-либо цепи нормального гемоглобина вследствие точечных мутаций генов. Известно около 300 вариантов HbA, имеющих в первичной структуре α- или β-цепи незначительные изменения. Некоторые из них практически не влияют на функции белка и здоровье человека, другие – вызывают значительные нарушения функции HbA и развитие заболеваний различной степени тяжести.
В аномальных гемоглобинах изменения могут затрагивать аминокислоты:
· находящиеся на поверхности белка;
· участвующие в формировании активного центра;
· аминокислоты, замена которых нарушает трехмерную конформацию молекулы;
· аминокислоты, замена которых изменяет четвертичную структуру белка и его регуляторные свойства.
Аномальные гемоглобины отличаются от HbA по первичной структуре, форме, величине заряда. При этом изменяются такие свойства как сродство к кислороду, растворимость, устойчивость к денатурации и др.
Примеры.
1. Серповидноклеточная анемия. Наследственное заболевание, связанное с заменой глутаминовой кислоты в 6-м положении (с N-конца) на валин в β-цепях молекулы гемоглобина S. Растворимость дезоксигемоглобина S значительно снижена. Его молекулы начинают «слипаться», образуя волокнистый осадок, который деформирует эритроцит, придавая ему форму серпа (полумесяца). Такие эритроциты плохо проходят через капилляры тканей, закупоривают сосуды и создают локальную гипоксию. Они быстро разрушаются и возникает гемолитическая анемия. Дети, гомозиготные по мутантному гену, часто умирают в раннем возрасте. Болезнь распространена в странах Южной Америки, Африки и Юго-Восточной Азии.
2. Гемоглобин М – в результате мутации в гене происходит замена в α- или β-цепи гистидина (в 7-м или 8-м положении) на тирозин. В результате этого Fe2+ окисляется в Fe3+ и образуется метгемоглобин, не способный связывать кислород. Развивается цианоз и гипоксия тканей.
Талассемии – наследственные заболевания, связанные с нарушением синтеза α- или β-цепей.
β-талассемии развиваются в результате снижения синтеза β-цепей. Проявляется после рождения, при этом в крови наряду с НbА появляется до 15 % НbА2 и 15-60 % HbF. Болезнь характеризуется гиперплазией и разрушением костного мозга, поражением печени, селезенки и сопровождается гемолитической анемией.
α-талассемии возникают при нарушении синтеза α-цепей. При полном отсутствии α-цепей наступает внутриутробная гибель плода, так как не образуется HbF, а тетрамеры γ4 обладают высоким сродством к кислороду и не способны выполнять транспортную функцию, что ведет к развитию тканевой гипоксии и к смерти вскоре после рождения.
Кооперативное взаимодействие
Взаимовлияние протомеров олигомерного белка друг на друга называется кооперативное взаимодействие.
В легких такое взаимодействие субъединиц гемоглобина повышает его сродство к кислороду и ускоряет присоединение кислорода в 300 раз. В тканях идет обратный процесс, сродство снижается и ускорение отдачи кислорода также 300-кратное.
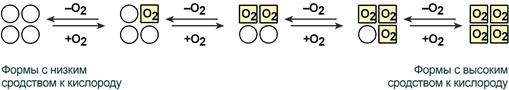
Схема кооперативного взаимодействия субъединиц гемоглобина
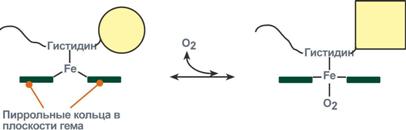
Объясняется такой феномен тем, что в легких при присоединении первой молекулы кислорода к железу (за счет 6-й координационной связи) атом железа втягивается в плоскость гема, кислород остается вне плоскости. Это вызывает перемещение участка белковой цепи и изменение конформации первого протомера. Такой измененный протомер влияет на другие субъединицы и облегчает связывание кислорода со второй субъединицей. Это меняет конформацию второй субъединицы, облегчая присоединение последующих молекул кислорода и изменение других протомеров.
Изменение формы субъединиц гемоглобина
при присоединении и отщеплении кислорода
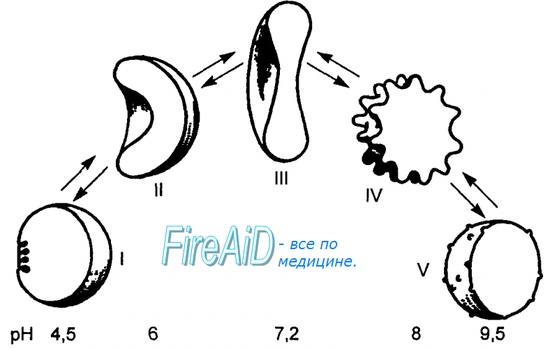
Дезоксиформа гемоглобина обозначается как Т-форма, напряженная (англ. tense), она обладает существенно более низким сродством к кислороду. Оксигенированная форма, или R-форма (англ. relaxed), обладает высоким сродством к кислороду.
Изменение рН среды
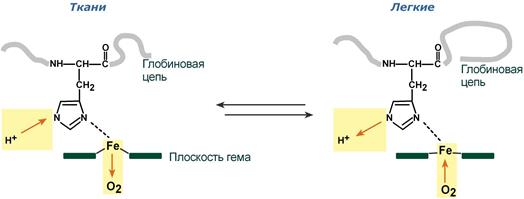
Влияние рН на сродство гемоглобина к кислороду носит название эффекта Бора. При закислении среды сродство снижается, при защелачивании – повышается.
При повышении концентрации протонов (закисление среды) в тканях возрастает освобождение кислорода из оксигемоглобина. В легких после удаления угольной кислоты (в виде СО2) из крови и одновременном увеличении концентрации кислорода высвобождаются ионы Н+ из гемоглобина.
Реакция взаимодействия кислорода с гемоглобином упрощенно имеет вид:

Изменение сродства гемоглобина к кислороду в тканях и в легких при изменении концентрации ионов H+ и О2 обусловлено конформационными перестройками глобиновой части молекулы. В тканях молекула О2 отрывается от железа и ионы водорода присоединяются к остаткам гистидина (глобиновой части), образуя восстановленный гемоглобин (H-Hb) с низким сродством к кислороду. В легких поступающий в больших количествах кислород “вытесняет” ион водорода из связи с остатком гистидина гемоглобиновой молекулы.
Механизм эффекта Бора
Роль 2,3-дифосфоглицерата
2,3-Дифосфоглицерат образуется в эритроцитах из 1,3-дифосфоглицерата, промежуточного метаболита гликолиза, в реакциях, получивших название шунт Раппопорта.
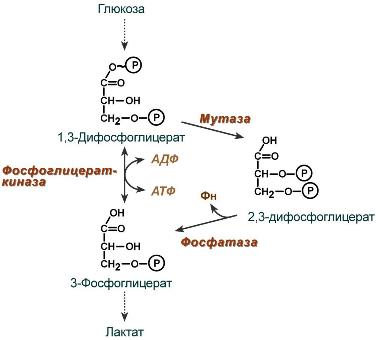
Реакции шунта Раппопорта
2,3-Дифосфоглицерат располагается в центральной полости тетрамера дезоксигемоглобина и связывается с β-цепями, образуя поперечный солевой мостик между атомами кислорода 2,3-дифосфоглицерата и аминогруппами концевого валина обеих β-цепей, также аминогруппами радикалов лизина и гистидина.
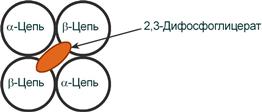
Расположение 2,3-дифосфоглицерата в гемоглобине
Функция 2,3-дифосфоглицерата заключается в снижении сродства гемоглобина к кислороду, что имеет особенное значение при подъеме на высоту и при нехватке кислорода во вдыхаемом воздухе. В этих условиях связывание кислорода с гемоглобином в легких не нарушается, так как концентрация его относительно высока. Однако в тканях за счет 2,3-дифосфоглицерата отдача кислорода возрастает в 2 раза.
Талассемия — это заболевание, из-за которого происходит нарушение синтеза гемоглобина. Название патологии дословно означает «анемия морского побережья». Заболевание наследуется по рецессивному типу, проявляется в результате генетических мутаций. Знания о том, что за болезнь талассемия, будут полезными при планировании семьи.
Этиология
Причиной талассемии являются точечные мутации или делеции в генах, кодирующих цепи гемоглобина. В результате это может привести к уменьшению синтеза или полному отсутствию одной из цепей в организме. Другая цепь образует неадекватные тетрамеры гемоглобина, что приводит к разрушению эритроцитов и гемолитической анемии.
Строение гемоглобина
Гемоглобин – белок содержащийся в эритроцитах, отвечающий за перенос кислорода к тканям и углекислого газа от них.
Гемоглобин (речь идёт о HbA) состоит из четырёх цепей: двух альфа-субъединиц и двух бета-субъединиц. Такой гемоглобин составляет 97% от общего содержания его в эритроцитах.
Оставшиеся 3% составляет гемоглобин HbA2, отличающийся по строению двух цепей: вместо бета-субъединиц у него дельта-субъединицы. HbA и HbA2 является вариантом нормы при их правильном соотношении.
Каждая из цепей гемоглобина связывается с его небелковой частью – гемом.
Так вот при талассемии нарушается синтез одной из цепей гемоглобина: либо альфа, либо бета. По этому принципу имеется классификация талассемии на:
- альфа-талассемию;
- бета-талассемию.
По степени тяжести выделяют талассемию:
- лёгкой степени;
- средней степени;
- тяжёлой степени.
Разновидности гемоглобина
Существуют физиологические виды гемоглобина и патологические.
К гемоглобину, который может быть в норме у человека, относится:
- HbP – примитивный гемоглобин, встречается в эмбрионе между 7 — 12-й неделей жизни;
- HbF – фетальный гемоглобин, содержит две альфа и две гамма-цепи, появляется через 12 недель внутриутробного развития, у взрослых его содержание составляет менее 1%;
- HbA – гемоглобин взрослых, доля составляет 97%, содержит две альфа и две бета-цепи;
- HbA2 – гемоглобин взрослых, доля составляет 2%, содержит две альфа и две дельта-цепи,
- HbO2 – оксигемоглобин, образуется при связывании кислорода в лёгких;
- HbCO2 – карбогемоглобин, образуется при связывании углекислого газа в тканях.
К патологическим формам гемоглобина относятся:
- HbS – гемоглобин, определяемый при серповидноклеточной анемии;
- MetHb – метгемоглобин, содержит трёхвалентный ион железа, когда в норме оно двухвалентное. Такая форма образуется при употреблении сульфаниламидов, нитратов, дефиците витамина С. Метгемоглобин не способен связывать кислород, в результате чего возникает гипоксия тканей;
- HbCO – карбоксигемоглобин, образуется при наличии избытка угарного газа во вдыхаемом воздухе. В крови он присутствует в небольших концентрациях, но его уровень может повышаться в зависимости от характеристики вдыхаемого воздуха.
HbS – вид гемоглобина, возникающего при мутации бета-цепи (одна аминокислота заменяется на другую). Он образуется у людей с серповидноклеточной анемией. Эритроциты, содержащие такой гемоглобин, долго не живут и быстро разрушаются, что хорошо в местах обитания малярийного плазмодия. У лиц с серповидноклеточной анемией имеется устойчивость к этому паразиту.
Почему разрушаются эритроциты?
Патогенез талассемии достаточно изучен. Первичные изменения начинаются с нарушенного синтеза гемоглобина, а именно белковых цепочек вещества. Известно, что гемоглобин, входящий в состав 90% массы эритроцитов, — единственное вещество, способное связывать молекулы кислорода и разносить их из легочной ткани по всему организму.
Его структура состоит из пигмента (гема), включающего железо, и набора из двух пар белковых цепочек. Их называют по типичному расположению аминокислот альфа- и бета-цепями. При нарушенном синтезе одного из типов полипептидов накапливается другой вид.
В результате разрушаются все клетки эритроцитарного ряда (сами эритроциты и их предшественники), из них выходит гемоглобин. Малокровие (анемия) развивается по гипохромному типу. Это подтверждается низким цветовым показателем.
«Виновниками» разрушений являются гены, ответственные за построение белковой части гемоглобина. Их мутация нарушает способность составлять необходимый набор аминокислот в цепи. Причину изменений связывают с возбудителем малярии — плазмодием. Доказаны его мутирующие способности. Талассемия по территории распространенности совпадает с эпидемическими зонами малярии.
Основные четыре цепочки белков, связывающие гем и влияющие на эритроциты
Дети могут получить болезнь путем наследования от родителей в двух видах:
- гомозиготном — ген-мутант передается от обоих родителей;
- гетерозиготном — ген болезни передается только от матери или отца, носителем может быть один из родителей.
Соответственно, называются формы талассемии.
При гетерозиготном носительстве выделяют:
- «немой» ген (α-th2);
- «манифестный» ген (α-th1).
Частота встречаемости
Талассемия наследуется аутосомно-рецессивно. Это значит, что поражает одинаково и мальчиков, и девочек. Рецессивный характер говорит о том, что дети больные талассемией появляются в тех семьях, где мама и папа оба являются носителями мутаций. Хотя они могут даже не догадываться о своём носительстве, так как симптомы порой бывают незаметны.
В среднем частота встречаемости составляет 1 на 100 000 человек и может изменяться в зависимости от региона. Бета-талассемия встречается чаще, чем альфа.
Альфа-талассемия
Существует 4 участка гена, которые кодируют синтез альфа-цепи гемоглобина. При наличии генетического дефекта (мутации) в них возникает альфа-талассемия. Выделяют несколько форм альфа-талассемии.
Гомозиготная
При ней нарушается синтез альфа-субъединиц на всех четырёх участках гена. В результате образуется гемоглобин, состоящий полностью из бета-цепей. Ещё он носит название гемоглобин Барта. Такой гемоглобин обладает высоким сродством к кислороду. Это означает, что он с трудом отдаёт кислород тканям. В результате возникает недостаток этого газа, что приводит к развитию сердечной недостаточности, отёкам, водянке и, как следствие, внутриутробной гибели плода.
Н-гемоглобинопатия
Н-гемоглобинопатия – форма, при которой нарушается синтез альфа-цепи на трёх участках гена. В результате избытка бета-субъединиц образуется HbH. У него также имеется достаточно высокое сродство к кислороду, он не стабилен, легко окисляется.
Присутствие такого гемоглобина в эритроцитах изменяет мембрану эритроцитов, сопровождается развитием гемолитической анемии, то есть усиленным распадом красных кровяных клеток. Проявляется такая форма альфа-талассемии к 1 году жизни в виде гемолитической анемии. Отмечаются признаки анемического синдрома, увеличение селезёнки. В анализах крови повышенные значения ретикулоцитов, что указывает на повышенную регенераторную способность костного мозга восстанавливать разрушенные клетки. Эритроциты гипохромные и похожи на мишень.
Малая альфа-талассемия
Малая альфа-талассемия – форма, при которой нарушен синтез 1 или 2 альфа-цепей. При такой форме отсутствуют тяжёлые клинические проявления. В крови может отмечаться лёгкая степень анемии (микроцитарная, гипохромная).
Несмотря на то, что нарушены 1 или 2 гена, отвечающих за синтез альфа-субъединиц, это не приводит к таким серьёзным нарушениям как, например, при бета-талассемии.
Прогноз заболевания
Гемотрансфузия массы эритроцитов совместно с медикаментозным лечением обеспечивают улучшение качества жизни пациента. Несмотря на это, подобная терапия полностью диагноз не устраняет, потому как обладает поддерживающим предназначением. Жизнь людей, пораженных тяжелым видом талассемии, обычно не превышает более 7 лет при условии регулярного прохождения процедур гемотрансфузии. Болезненное деформирование внутренних органов отличается прогрессированием, что приносит ребенку страдания.
Бета-талассемия
Бета-талассемия возникает при нарушении синтеза бета-субъединиц. Вследствие мутаций образуется гемоглобин, в котором бета-цепи замещаются на альфа. В эритроцитах образуются альфа-тетрамеры. Ретикулоэндотелиальная система удаляет их из красных кровяных клеток, они повреждаются, и происходит гемолиз.
В результате мутаций в гене бета-цепи различают большую и малую бета-талассемии.
Малая бета талассемия
Малая (минорная) бета-талассемия – гетерозиготная форма, характеризующаяся лёгким течением.
В лабораторных анализах:
- в норме или повышено количество сидеробластов;
- гипохромная микроцитарная анемия;
- анизоцитоз – разные эритроциты по размеру;
- пойкилоцитоз – эритроциты разной формы;
- повышено количество ретикулоцитов;
- железо в норме;
- повышенные уровни непрямого билирубина.
У больных малой формой бета-талассемии отмечается повышение уровня HbA2 (до 6%, норма до 3%) и HbF (до7%, в норме – менее 1%).
Малую форму бета-талассемии зачастую путают с железодефицитной анемией, ошибочно лечат препаратами железа. Для дифференциальной диагностики этих двух состояний необходимо проводить оценку уровня железа в крови. При бета-талассемии оно будет в пределах нормы.
Большая талассемия
Большая талассемия (анемия Кули) – гомозиготная, считается тяжёлой прогрессирующей формой бета-талассемии. Как и другие формы талассемии, она проявляется бледностью кожи, увеличением размеров селезёнки. Они возникают к 1 году ребёнка. Особенностью анемии Кули являются изменения костей: выступающие скулы, узкие глазные щели, квадратный череп, плоская переносица. У таких детей физическое развитие на низком уровне.
В лабораторных анализах отмечается:
- повышенное содержание сидеробластов;
- низкие показатели MCV, МСН, МСНС, свидетельствующие о гипохромной микроцитарной анемии;
- анизоцитоз;
- пойкилоцитоз – в виде мишени, шизоциты;
- базофильная пунктация эритроцитов;
- увеличение осмотической резистентности эритроцитов;
- увеличение неконъюгированного билирубина;
- избыточное содержание железа, вплоть до отложения его в органах (гемосидероз).
Для большой бета-талассемии характерно увеличение содержания в крови фетального (HbF) гемоглобина до 70%, что подтверждается при проведении электрофореза.
Сидеробласты – их ещё по-другому называют эритробластами. Они содержат в цитоплазме негемоглобиновое железо в виде гемосидерина и ферритина.
Немного истории
Болезнь была впервые описана в 1925 году американскими врачами-педиатрами, которые при лечении эмигрантов из Италии выявили одинаковую клиническую картину у детей с тяжелым малокровием, увеличением печени и селезенки, изменениями костей.
Затем появились работы, описывающие более легкое течение болезни у взрослых пациентов. Термин «талассемия» предложили в 1936 году. Дословно он означает «болезнь морского побережья». Высказана мысль о связи патогенеза заболевания с нарушением синтеза глобиновых цепочек.
Общие клинические признаки талассемии
- Гемолитическая анемия приведёт к бледности, вялости и иногда даже к пожелтению кожного покрова.
- Живот будет увеличен за счёт спленомегалии (увлечение селезёнки).
- При некоторых формах талассемии отмечаются костные деформации.
- У детей, больных талассемией наблюдается задержка физического развития, обусловленная нехваткой кислорода для роста и развития тканей.
- Из-за повышенного всасывания железа в кишечнике и частых переливаний крови наблюдается повышение этого элемента в крови, он откладывается в органах и тканях (например, сердце, печень), нарушая их работу.
Возможные осложнения и прогноз
При тяжелой форме патологии в тканях накапливается гемосидерин. У больных развивается цирроз печени, диабет. Гемосидероз миокарда приводит к гибели пациента.
При гемоглобинозе Н проводят удаление селезенки. Из-за того, что больные склонны к развитию инфекционных осложнений, им необходимо проходить вакцинацию против пневмококковых инфекций.
Прогноз большинства разновидностей гомозиготной талассемии неблагоприятный. Больные умирают в младенческом либо молодом возрасте. При тяжелых формах патологии продолжительность жизни больного зависит от своевременной терапии и предупреждения гемосидероза.
При гетерозиготной форме талассемии качество жизни пациентов не страдает. Трансплантация костного мозга улучшает прогноз.
Диагностика талассемии
При диагностике талассемии нельзя ориентироваться только на одну лишь клинику, тем более, что она не всегда может быть, например, при лёгких формах. Поэтому для точной диагностики необходимо «заглянуть» внутрь организма и оценить процессы, происходящие в нём. В этом помогут лабораторные исследования крови.
На анемию укажут низкий уровень гемоглобина и эритроцитов крови. При микроскопическом исследовании мазка крови будет замечена форма красных кровяных клеток и содержание гемоглобина в них. То есть подтвердится микроцитарный гипохромный характер анемии.
Электрофорез гемоглобина позволит оценить, как изменился его фракционный состав (соотношение HbA, HbA2, HbF между собой).
Ультразвуковое исследование позволит увидеть увеличение размера селезёнки.
Рентгеновское исследование используется для изучения костных деформаций.
Пункция костного мозга позволит оценить процессы кроветворения и сделать заключение о выраженности процессов.
Главным исследованием, которое может подтвердить наличие талассемии, является молекулярно-генетический анализ. Выявленная мутация гена альфа или бета-цепей свидетельствует о талассемии.
Биохимическое исследование крови позволит определить ряд показателей, которые могут повышаться при талассемии, например, непрямой билирубин. Оценка метаболизма железа в организме позволит провести дифференциальную диагностику между железодефицитной анемией и талассемией.
Перед тем как приступить к лечению, необходимо провести генетическое исследование, чтобы определить тип мутации и дальнейший прогноз развития заболевания.
Профилактические меры
Для будущих мам, находящихся в положении, проводится полная дородовая диагностика, обследования генетического характера
Профилактика бета-талассемии основана на предотвращении зачатия и появления на свет больных детей. Выполняется это несложно посредством простого исследования крови и процесса скрининга. Носителей заболевания предостерегают об угрозе связи с партнерами, пораженными таким же диагнозом.
Для снижения опасности рождения больного ребенка у пары с талассемией берется кровь на исследования различного рода. Для будущих мам, находящихся в положении, проводится полная дородовая диагностика, обследования генетического характера. Исследования ДНК и тесты ворсинок позволяют с начала беременности выявить гемоглобинопатию.
Чтобы оградить своих детей от любых заболеваний будущей мамочке необходимо всегда следить за своим здоровьем, а в случае наличия бета-талассемии, важное значение будет играть тщательный подход к выбору партнера.
На сегодняшний день заболевание считается неизлечимым. Новорожденный с легким типом бета-талассемии может прожить полноценную жизнь без серьезных последствий. Однако гарантировать непосредственно такую форму недуга современные ученые возможности не имеют. В особенности задуматься о деторождении нужно парам с гомозиготной наследственностью.
Лечение талассемии
Талассемия – наследственное заболевание, поэтому существует только симптоматическая терапия.
Лечение талассемии будет зависеть от формы. При лёгких формах терапия не проводится. При необходимости проводят переливание крови. Пациентам в бета-талассемией необходимо следить за уровнем железа в крови, в случае его избытка им должны быть назначены железо-связывающие лекарственные средства.
Пациентам с тяжёлыми формами назначается переливание крови в комбинации с препаратами, выводящими железо из организма. Гемоглобин поддерживают на уровне 100 г/л.
Дефероксамин относится к тем препаратам, который способен образовывать комплексы с железом и выводить его с мочой. Самостоятельно ничего не принимайте, все лекарственные средства только по назначению врача.
Если облегчение симптомов после переливания крови не наступает, может быть проведена операция по удалению селезёнки.
Профилактика
Так как заболевание талассемия является генетически обусловленным, то первичная профилактика включает в себя дородовую диагностику. Если оба родителя болеют талассемией, необходимо провести исследование плода. Иногда врач рекомендует прерывание беременности.
При обследовании беременных применяют фетоскопию, амниоцентез (под контролем ультразвука). В дальнейшем проводят генетическое обследование плода.
Родителям, которые имеют родственников, страдающих талассемией, нужно обратиться перед планированием беременности к генетику. В случае необходимости назначают генетическое обследование.